Arvutikeemia — definitsioon, molekulide modelleerimine ja rakendused
Avasta arvutikeemia: molekulide modelleerimine, ennustused ja rakendused uute ravimite ning materjalide kavandamisel — täpsed ja efektiivsed meetodid.
Arvutikeemia on keemia haru, mis kasutab keemiliste probleemide lahendamiseks arvutiteadust. Need programmid arvutavad molekulide ja tahkete ainete struktuure ja omadusi. Arvutikeemia täiendab tavaliselt keemiliste katsete abil saadud teavet. Sellega saab ennustada keemilisi nähtusi, mida ei ole veel täheldatud. Seda kasutatakse laialdaselt uute ravimite ja materjalide kavandamisel.
Arvutikeemia abil saab ennustada struktuuri (st molekuli aatomite eeldatavat asendit), absoluutseid ja suhtelisi (vastastikmõju) energiaid, elektronlaengute jaotusi, dipoole ja kõrgemaid multipoolmomente, võnkesagedusi, reaktiivsust või muid spektroskoopilisi suurusi ning ristlõikeid kokkupõrgeteks teiste osakestega.
Arvutikeemia uurib nii staatilisi kui ka dünaamilisi süsteeme. Kõikidel juhtudel kasvab uuritava süsteemi suuruse kasvades ka kasutatav arvutiaeg ja muud ressursid (näiteks mälu ja kettaruum). See süsteem võib olla üks molekul, molekulide rühm või tahke aine. Arvutikeemia meetodid varieeruvad väga täpsetest kuni väga ligikaudsete meetoditeni. Väga täpsed meetodid on tavaliselt teostatavad ainult väikeste süsteemide puhul.
Pildigalerii
8 Pildid


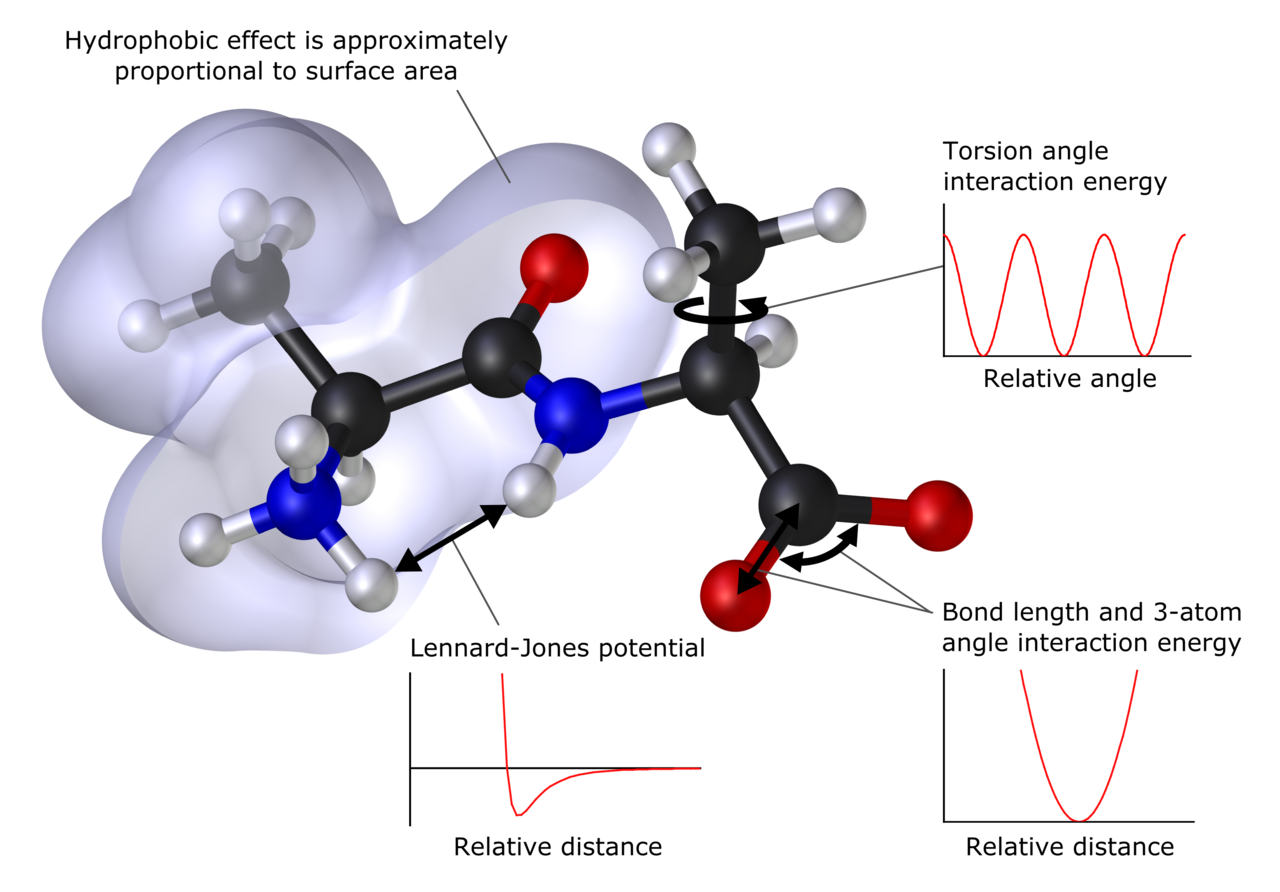


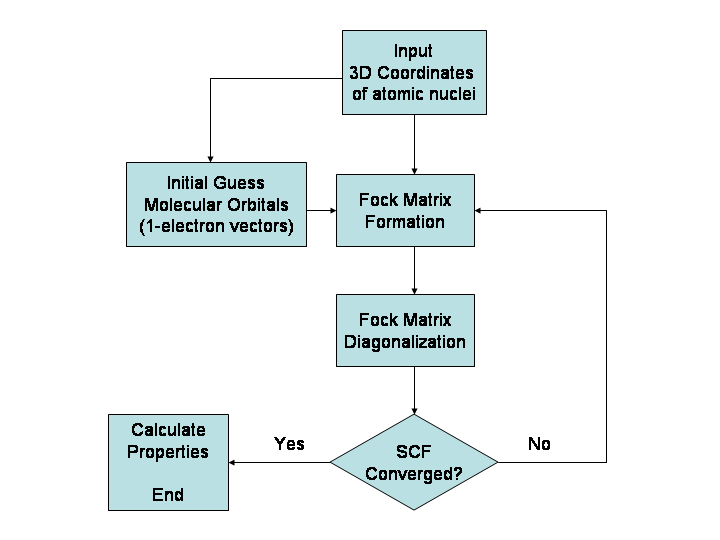
Peamised meetodid
- Ab initio (esialgsetest eeldustest lähtuvad) meetodid: põhinevad kvantmehaanikal ja ei kasuta empirilisi parameetreid. Näited: Hartree–Fock (HF) ja täiendusmeetodid (MP2, CCSD(T)). Need annavad kõrge täpsuse väikestele molekulidele, kuid on arvutuslikult kallid.
- Tihendusfunktsiooni tiheduse lähenemine (DFT): praktiline ja laialt kasutatav kvantmehaaniline meetod, mis tasakaalustab täpsuse ja arvutuskoormuse. Sobib nii orgaaniliste molekulide kui ka tahkete ainete uurimiseks.
- Semiempirilised meetodid: kasutavad mõningaid empirilisi parameetreid, et vähendada arvutusaega. Sobivad suurte molekulide kiireks hindamiseks, kuid täpsus võib varieeruda.
- Molekulaarmehaanika (MM): modelleerib molekule klassikaliste potentsiaalide (force field) abil — ei hõlma otseselt elektronide kvantmehhaanikat. Hea suurtel biomolekulidel (valkudel, nukleiinhapetel) ja polümeeridel.
- Molekulaarne dünaamika (MD): simuleerib aatomite ja molekulide liikumist aja funktsioonina, võimaldades uurida termodünaamilisi ja kineetilisi omadusi. MD võib olla klassikaline (force field) või kvantmehhaanika+MM hübriid (QM/MM).
- Monte Carlo-meetodid: statistilised simulatsioonid, mida kasutatakse peamiselt termodünaamiliste proportsioonide ja faasikäitumise hindamiseks.
Mis tüüpi tulemusi arvutikeemiast saada on võimalik?
- Geomeetriaoptimeerimine — aatomite tasakaalupositsioonid ja sidemete pikkused/nurgad.
- Energiate arvutused — reaktsiooniteed, aktiveerimisenergiad, võrreldavad energiad eri konfiguratsioonide vahel.
- Spektroskoopilised parameetrid — vibratsioonisagedused, NMR-keemilised tõuked, UV/VIS- ja IR-eksitused.
- Elektronilise struktuuri omadused — orbitaalide energiad, elektronjaotused, dipoolmomendid.
- Termodünaamilised ning kineetilised protsessid — faasidiagrammid, reaktsioonikiirused, barjäärid.
- Ristsündmuste ja kokkupõrgete ristlõiked — põhimõtteliselt oluliseks elektrontransportis ja spektroskoopias.
Rakendused
- Ravimite disain — ligandite ja retseptorite interaktsioonide kiire hindamine, virtuaalne ekraanimine ja molekulaarse optimeerimise abivahendid.
- Materjaliteadus — uute pooljuhtide, katalüsatorite, akumaterjalide ja polümeeride omaduste ennustamine ning struktuuride kavandamine.
- Katalüüs ja reaktsioonimehhanismid — reaktsiooniteede modelleerimine, aktiveerimisbarjääride leidmine, katalüütiliste protsesside optimeerimine.
- Spektroskoopia — eksperimendi tulemuste tõlgendamine ja ootuspäraste spektrite ennustamine enne mõõtmisi.
- Keskkonnakeemia — saasteainete lagunemisteede modelleerimine ja teratogeensuse/võimalike riskide hindamine.
Piirangud ja vead
- Arvutikeemia täpsus sõltub mudeli tasemest — lihtsamad meetodid võivad anda eksitavaid tulemusi tundlike suuruste jaoks.
- Arvutuslik keerukus kasvab kiiresti süsteemi suuruse ja kasutatava teooria taseme tõustes (nn eksponentsiaalne või polünoomne skaala sõltuvalt meetodist).
- Paljud meetodid eeldavad lähteolekuid (näiteks alggeomeetria, baasfunktsioonide valik), mis võivad mõjutada lõpptulemust.
- Süsteemide reaalsete tingimuste (lahusti mõju, temperatuur, rõhk) adekvaatne modelleerimine nõuab täiendavaid meetodeid (nt QM/MM, polariseerivad force field’id või Monte Carlo simulatsioonid).
Tarkvara ja ressursid
- Levinud tarkvarapaketid: Gaussian, ORCA, NWChem, VASP, Quantum ESPRESSO, GROMACS, AMBER, LAMMPS jt. Valik sõltub uuritavast probleemist (kvantmehhaanika vs klassikaline MD, tahke aine vs molekulaarne süsteem).
- Arvutusressursid: tihti vajalikud on kõrgjõudlusarvutid (HPC) või GPU-põhised lahendused suuremahuliste simulatsioonide jaoks.
- Andmebaasid ja töövood: ligipääs molekulaarsetele andmebaasidele, parameetrite komplektidele (force field’id) ja töövoogudele lihtsustab korduvaid ülesandeid ning automatiseerimist.
Praktilised nõuanded
- Alusta alati lihtsamatest meetoditest ja võrrelge tulemusi järk-järgult täpsemate meetoditega, kui vaja.
- Kontrolli tulemuste sõltuvust baasfunktsioonide ja parameetrite valikust.
- Kasutage eksperimente kui võimalikaks kinnitusena — arvutused on tihti kõige tugevamad koos laborikatsetega.
- Pööra tähelepanu staatilise ja dünaamilise simulatsiooni piirangutele (ajaastmed, simulatsiooni pikkus, algtingimused).
Tulevikusuundumused
Arvutikeemiasse jõuavad järjest enam masinõppe- ja andmepõhised meetodid, mis kiirendavad ennustuste tegemist ja aitavad leida uusi materjale või ravimikandidaate. Samuti on kiire arendus kvantarvutuse valdkonnas — kuigi praktilised rakendused on alles algusjärgus, võivad need tulevikus muuta keerukate kvantmehhaaniliste probleemide lahendamist.
Kokkuvõte
Arvutikeemia on mitmekülgne ja praktiline distsipliin, mis ühendab kvantmehaanika, statistika ja arvutusmeetodid, et mõista ja ennustada keemilisi protsesse. See on väärtuslik täiendus eksperimentaalsele keemiale ning mängib võtmerolli ravimite, materjalide ja katalüsaatorite arendamisel. Õige meetodi ja ressursi valik ning tulemuste kriitiline hindamine on edu aluseks.

Seotud leheküljed
- Bioinformaatika
- Statistiline mehaanika
Küsimused ja vastused
K: Mis on arvutuslik keemia?
V: Arvutikeemia on keemia haru, mis kasutab keemiliste probleemide lahendamiseks arvutiteadust. Seda saab kasutada molekulide ja tahkete ainete struktuuride ja omaduste arvutamiseks, seni vaatlemata keemiliste nähtuste ennustamiseks ning uute ravimite ja materjalide kavandamiseks.
K: Milliseid süsteeme arvutuslik keemia uurib?
V: Arvutikeemia uurib nii staatilisi kui ka dünaamilisi süsteeme. Süsteem võib olla üks molekul, molekulide rühm või tahke aine.
K: Millist teavet saab arvutuslik keemia anda?
V: Arvutikeemia võib anda sellist teavet nagu struktuur (aatomite asukohad), absoluutsed ja suhtelised energiad, elektronlaengute jaotused, dipoolid ja kõrgemad multipoolmomendid, võnkesagedused, reaktsioonivõime või muud spektroskoopilised suurused ning ristlõiked kokkupõrkete puhul teiste osakestega.
K: Kui täpsed on arvutuslikus keemias kasutatavad meetodid?
V: Arvutikeemias kasutatavate meetodite täpsus ulatub väga täpsest kuni väga ligikaudse täpsuseni. Väga täpsed meetodid on tavaliselt teostatavad ainult väikeste süsteemide puhul.
K: Kuidas täiendab arvutuslik keemia eksperimentaalseid andmeid?
V: Arvutikeemia täiendab tavaliselt keemiliste katsete abil saadud teavet. Selle abil saab ennustada tulemusi, mida ei ole veel eksperimentaalselt täheldatud.
K: Kas uuritava süsteemi suurus mõjutab seda, kui palju arvutiaega on vaja?
V: Jah - kui uuritava süsteemi suurus kasvab, siis kasvab ka analüüsiks vajalik arvutiaeg ja ressursid, näiteks mälu ja salvestamiseks vajalik kettaruum.
Seotud artiklid
Autor
AlegsaOnline.com Arvutikeemia — definitsioon, molekulide modelleerimine ja rakendused Leandro Alegsa
URL: https://et.alegsaonline.com/art/22297