Energiatasemed kvantmehaanikas: aatomite ja molekulide elektronide ülevaade
Sügav ülevaade energiatasemetest kvantmehaanikas: aatomite ja molekulide elektronide konfiguratsioonid, degeneratsioon, potentsiaal ja spektrid praktiliste näidetega.
See artikkel käsitleb orbitaalide (elektronide) energiatasemeid. Ühendite energiatasemete kohta vt keemiline potentsiaal.
Energiatasemed defineeritakse kvantmehaanikas tavaliselt kui aatomi või molekuli elektronide võimalikud energiad, st potentsiaalse + kineetilise energia kindlad väärtused, mida süsteem võib omada. Kuna Kvantmehaaniline süsteem ei saa võtta suvalist energiat, vaid ainult diskreetseid väärtusi (seotud olekute puhul), räägitakse sageli diskreetsetest energiatasemetest; samas on olemas ka pidev spekter vabanemis- või ionisatsioonienergiate puhul.
Pildigalerii
2 Pildid
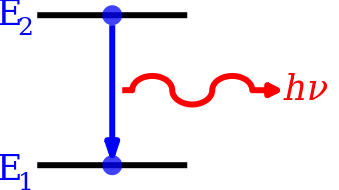
Mis määrab energiataset?
Peamised tegurid, mis määravad elektronide energiatasemed, on:
- tuuma laeng (mürganõudlus) ja sellest tulenev Coulomb'i potentsiaal;
- elektronide omavaheline tõrje ja korrelatsioonid;
- kvantarvud ja orbitaalide kujud (n, l, m, s);
- spin-orbiidi interaktsioon (fine-ehitis) ja hüperfine-ehitus (tuuma voolud ja magnetmomendid);
- keskkonna välismõjud: välised elektri- ja magnetväljad (Stark- ja Zeemani efekt), rõhk, temperatuur jms.
Aatomite energiatasemed
Aatomimudeli lihtsaim näide on vesinikuaatom, mille jaoks saab Schrödingeri võrrandist analüütilised lahendid: energiatasemed sõltuvad peamiselt peamisest kvantarvust n ja on diskreetsed. Küll aga mitmeelektronilistes aatomites mängivad olulist rolli ka elektronidevaheline tõrje ja tõhus tuumalaeng (effective nuclear charge), mistõttu orbitaalide energiaperioodid (nt 2s vs 2p) ei võrdu enam hulga degeneratsiooniga, nagu happe vesiniku puhul.
Olulised mõisted aatomite puhul:
- Kvantarvud: n (peamine), l (orbitaalne), m (magnetiline) ja s (spin) määravad orbitaalide kuju ja orientatsiooni;
- Pauli printsiip: igal aatomil ei saa olla kahte elektroni, millel on kõik kvantarvud identsed — see viib elektronide paigutumiseni erinevatesse orbitaalidesse;
- Degeneratsioon: samal energiatasemel olevad erinevad kvantmehaanilised olekud on degeneratiivsed energiatasemed;
- Hund'i reeglid ja elektronide konfiguratsioon: elektronid eelistavad täita elektronide elektronispinnid maksimaalse spiniga ning järjestus orbitaalides sõltub energiate järjestusest — vaata ka elektronide konfiguratsiooniga seotud selgitusi.
Molekulide energiatasemed
Molekulide puhul tuleb arvestada nii elektronide kui tuumade omavahelist dünaamikat. Sageli eeldatakse Born–Oppenheimer lähenemist, kus elektronid liiguvad palju kiiremini kui tuumad, mistõttu elektronide energiatasemed arvutatakse tuumade fikseeritud konfiguratsiooni juures ja saadud pinnad (potential energy surfaces) määravad molekulaarse vibratsiooni- ja rotatsioonispektri.
Molekulides on olulised nüansid:
- Molekulaarsed orbitaalid: aatomiorbitaalide kombineerimisel tekivad siduvad (bonding) ja mittesiduvad (antibonding) orbitaalid — nende energiate erinevus määrab sideme tugevuse;
- HOMO ja LUMO: kõrgeim täidetud ja madalaim täitmata molekulaarne orbitaal on olulised reaktsioonivõime ja optiliste üleminekute jaoks;
- Vibratsioonilised ja rotaarsed tasandid: iga elektroniline olek kannab endaga kaasas arvukalt vibratsioonilisi ja rotaarseid alamtasandeid (termiline populatsioon ja Franck–Condon faktorid määravad üleminekute intensiivsust);
- Potentsiaali pinnad ja reaktiivsus: elektronseisundite energia sõltub tuumade kaugusest — keemilised reaktsioonid liiguvad mööda nende pindade ristumiskohti (esimene ja teine järku üleminekusuunad).
Spektraalsed üleminekud ja reeglid
Energiatasetest saab palju teavet elektromagnetilise kiirguse neeldumise ja emissiooni kaudu. Üleminekute toimumiseks kehtivad valikureeglid (näiteks muutus orbitaalse kvantarvu l = ±1 dipoolüleminekul). Lisaks mõjutavad intensiivsust ja joonekuju:
- Franck–Condon faktorid (molekulaarsete vibratsioonide kattumine elektroonses üleminekus);
- lahutusvõime, looduslik ja termiline laienemine (joone laius: eluiga vs laienemine);
- mitteradiaatiivsed protsessid (intersüsteemne ristsiire, sisemine ümberjaotumine), mis lühendavad eluiga ja muudavad emissiooni tõhusust.
Lisamõjud ja täiendavad nähtused
- Õõtsumine ja kvantne tunnel: kitsa barjääri puhul võivad elektronid „tunnelduda” läbi barjääri — oluline näiteks molekulaarsetes reaktsioonides ja tahkistes;
- Väli-efektid: välised magnet- ja elektriväljad lõhuvad degeneratsiooni (Zeemani ja Stark'i efektid) ja muudavad tasemete järjestust;
- Korrelatsioon ja paljuelektronilised arvutused: täpsete tasemete leidmiseks tuleb arvestada elektronide korrelatsiooniga — lihtsad Hartree–Fock meetodid annavad sageli lähendi, kuid täpsemad meetodid (nt konfiguratsioonisuhtelised, mitmereferentsilised meetodid) on vajalikud komplekssemate süsteemide jaoks;
- Solidi-olek ja bändistruktuur: suurtes süsteemides (metallid, pooljuhid) lähevad aatomitasemed laiali ja moodustavad elektroni- ja augubändid; elektronide täituvus bändides määrab materjali elektrilised omadused.
Mõned praktilised mõõtmis- ja rakendusvaldkonnad
- elektron- ja optilised spektrid (oksüdeerimine, absorptsioon-, luminesentsspektroskoopia);
- fotoelektronspektroskoopia (PES/UPS) tuvastamaks orbitaalide energiaid ja tööfunktsiooni;
- kvantkeemilised arvutused materjalide ja katalüsaatorite disainis;
- laseritega juhitavad üleminekud ja kvantsüsteemide manipuleerimine (kvantinformaatika, kvantpunktid).
Lõpetuseks: energiatasemed kvantmehaanikas annavad sisulise kirjelduse elektronide võimalikest olekutest aatomites ja molekulides ning on aluseks keemilisele sidemele, spektroskoopiale ja paljudele kaasaegsetele tehnoloogiatele. Tegurid nagu elektronidevaheline tõrje, orbitaalne kuju, spin-orbiidi vastasmõjud ja keskkonna välismõjud võivad energiaid nihutada, lõhkumata aga põhimõtteliselt kvantiseeritud olemust — see ongi põhjus, miks energiatasemed on nii fundamentaalsed nii teoorias kui rakendustes.
Aatomid
Omane energiatase
Orbitaalriigi energiatase
Oletame, et elektron on antud aatomi orbitaalis. Selle oleku energia on peamiselt määratud (negatiivse) elektroni ja (positiivse) tuuma elektrostaatilise koostoime kaudu. Elektroni energiatasemed tuuma ümber on antud järgmiselt: :
E n = - h c R ∞ Z n 2{\displaystyle2 E_{n}=-hcR_{\infty }{\frac {Z^{2}}{n^{2}}}}\ }  ,
,
kus R ∞ {\displaystyle R_\infty }\  on Rydbergi konstant (tavaliselt vahemikus 1 eV kuni 103 eV), Z on aatomi tuuma laeng, n {\displaystyle n\ }
on Rydbergi konstant (tavaliselt vahemikus 1 eV kuni 103 eV), Z on aatomi tuuma laeng, n {\displaystyle n\ } on peamine kvantarv, e on elektroni laeng, h {\displaystyle h}
on peamine kvantarv, e on elektroni laeng, h {\displaystyle h} on Plancki konstant ja c on valguse kiirus.
on Plancki konstant ja c on valguse kiirus.
Rydbergi tasemed sõltuvad ainult põhikvantarvust n {\displaystyle n\ }. .
Peenstruktuuri lõhenemine
Peenstruktuur tuleneb relativistlikest kineetilise energia korrektsioonidest, spin-orbiti sidemest (elektroni spinni ja liikumise ning tuuma elektrivälja vaheline elektrodünaamiline vastastikmõju) ja Darwini terminist (s-kujuliste elektronide kontakttermiline vastastikmõju tuuma sees). Tüüpiline suurus10 - 3{\displaystyle 10^{-3}} eV.
eV.
Hüperfiinne struktuur
Spin- tuuma-spin-kohtumine (vt hüperfiinstruktuur). Tüüpiline suurus10 - 4{\displaystyle 10^{-4}} eV.
eV.
Elektroni elektrostaatiline vastastikmõju teiste elektronidega
Kui aatomi ümber on rohkem kui üks elektron, tõstavad elektron-elektron-interaktsioonid energiataset. Neid vastastikmõjusid jäetakse sageli tähelepanuta, kui elektronide lainefunktsioonide ruumiline kattuvus on väike.
Välistest väljadest tingitud energiatasemed
Zeemani efekt
Interaktsioonienergia on: U = - μ B {\displaystyle U=-\mu B}  koos μ = q L / m 2{\displaystyle \mu =qL/2m}
koos μ = q L / m 2{\displaystyle \mu =qL/2m} 
Zeemani efekt, mis võtab arvesse spinni
See võtab arvesse nii orbitaalsest nurkmomendist tulenevat magnetilist dipoolmomenti kui ka elektroni spinnist tulenevat magnetilist momenti.
Relativistlike efektide tõttu (Diraci võrrand) on elektroni spinnist tulenev magnetiline moment μ = - μ B g s {\displaystyle \mu =-\mu _{B}gs} , kusjuures g {\displaystyle g}
, kusjuures g {\displaystyle g} on güromagnetiline tegur (umbes 2). μ = μ l + g μ s {\displaystyle \mu =\mu _{l}+g\mu _{s}}
on güromagnetiline tegur (umbes 2). μ = μ l + g μ s {\displaystyle \mu =\mu _{l}+g\mu _{s}}  Vastasmõju energia saab seega U B = - μ B = μ B B ( m l + g m s ) {\displaystyle U_{B}=-\mu B=\mu _{B}B(m_{l}+gm_{s})}
Vastasmõju energia saab seega U B = - μ B = μ B B ( m l + g m s ) {\displaystyle U_{B}=-\mu B=\mu _{B}B(m_{l}+gm_{s})} .
.
Stark mõju
Vastasmõju välise elektriväljaga (vt Starki efekt).
Molekulid
Molekuli energiaseisund, s.t molekulaarse Hamiltiani omastase, on elektroonilise, võnke-, pöörlemis-, tuuma- ja translatsioonikomponendi summa, nii et:
E = E e l e k t r o n i k + E v i b r a t i o n a l + E r o t a t i o n a l + E n u k l a a r + E t r a n s l a t i o n a l {\displaystyle E=E_{\mathrm {electronic} }+E_{\mathrm {vibratsiooniline} + E_{\mathrm {vibratsiooniline} }+E_{\mathrm {rotatsiooniline} + E_{\mathrm {rotatsiooniline} }+E_{\mathrm {nuclear} + E_{\mathrm {nuclear} }+E_{\mathrm {translatsiooniline} }\,} 
kus E e l e k t r o n i k {\displaystyle E_\mathrm {electronic} }} on molekuli elektroonilise molekulaarhamiltonija omaväärtus (potentsiaalse energiapinna väärtus) molekuli tasakaalugeomeetrias.
on molekuli elektroonilise molekulaarhamiltonija omaväärtus (potentsiaalse energiapinna väärtus) molekuli tasakaalugeomeetrias.
Molekulaarsed energiatasemed on tähistatud molekulaarsete terminite sümbolitega.
Nende komponentide erienergiad varieeruvad sõltuvalt konkreetsest energiast ja ainest.
Molekulaarfüüsikas ja kvantkeemias on energiatase seotud kvantmehaanilise oleku kvantitud energia.
Kristallilised materjalid
Kristallilisi materjale iseloomustavad sageli mitmed olulised energiatasemed. Kõige olulisemad neist on valentsusriba ülemine osa, juhtivusriba alumine osa, Fermi energia, vaakumitase ja kristallide võimalike defektide energiatasemed.
Seotud leheküljed
Küsimused ja vastused
K: Mis on orbitaalenergia tasemed?
V: Orbitaalide energiatasemed on aatomi elektronide potentsiaalse energia erinevad olekud, mis on määratletud kvantitatiivselt mõõdetava energiaspektrina.
K: Miks võib kvantmehaaniline süsteem olla ainult teatud seisundites?
V: Kvantmehaaniline süsteem võib olla ainult teatud olekutes, sest energiatasemed on kvantitud, st ainult teatud energiatasemed on võimalikud.
K: Mis on degenereerunud energiatasemed?
V: Degenereerunud energiatasemed on energiatasemed, mida saadakse rohkem kui ühest kvantmehaanilisest olekust.
K: Millal on potentsiaalne energia nulliks?
V: Potentsiaalne energia on tavaliselt nulliks seatud lõpmatuseni.
K: Mis on mõiste energiatase kõige tavalisem kasutusviis?
V: Enamasti kasutatakse terminit energiatase seoses elektronide konfiguratsiooniga aatomites või molekulides.
K: Mis määrab aatomite ja molekulide energiatasemed?
V: Kõige olulisemaid tegureid, mis määravad aatomite ja molekulide energiatasemeid, käsitletakse artikli järgmistes osades.
K: Kas on juhtumeid, kus energiaspekter ei ole kvantitud?
V: Jah, on juhtumeid, kus energiaspekter ei ole kvantitud, mida nimetatakse pidevaks spektriks. Orbitaalide energiatasemete kontekstis on energiaspekter siiski kvantitud.
Seotud artiklid
Autor
AlegsaOnline.com Energiatasemed kvantmehaanikas: aatomite ja molekulide elektronide ülevaade Leandro Alegsa
URL: https://et.alegsaonline.com/art/31417