Osteogenesis imperfecta (hapra luuhaigus): põhjused, sümptomid ja tüübid
Osteogenesis imperfecta (hapra luuhaigus) — geneetilised põhjused, sümptomid ja tüübid: luumurrud, kollageeni nõrkus, kuulmislangus ja hambaprobleemid ning elukvaliteedi nõuanded.
Osteogenesis imperfecta on geneetiline häire, mida tuntakse ka tavapäraselt hapra luuhaiguse nime all. Haigus mõjutab peamiselt luude tugevust, sest puuduliku või väärarenguga kollageeni tõttu muutuvad luud kergemini murduvaks. See seisund avastati meditsiinis juba 19. sajandil (Vrolik, 1854) ja tänapäeval teatakse mitmeid haiguse alaliike ja päriluse vorme.
Pildigalerii
10 Pildid







Põhjused ja pärilikkus
Enimlevinud põhjused on mutatsioonid geenides, mis kodeerivad I tüüpi kollageeni ehitust. Kõige sagedasemad mõjutatud geenid on COL1A1 ja COL1A2. Tavaliselt on osteogenesis imperfecta autosomaalselt domineeriv, mis tähendab, et haigust põhjustav muutus ühest geenikopiast võib põhjustada haiguse avaldumist. Samas on kirjeldatud ka teisi pärimismustreid (nt autosomaalne retsessiivne), ning mõnel juhul tekib mutatsioon uue (de novo) muutusena inimesel, kelle vanematel haigust polnud.
Sümptomid ja tunnused
Sümptomite raskusaste võib väga varieeruda — alates kergetest juhtudest, kus esinevad vaid harvad luumurrud ja normaalse pikkuse säilimine, kuni raskete vormideni, mis põhjustavad korduvaid murranguid, luude deformeerumist ja eluohtlikke tüsistusi. Tavalisemad tunnused on:
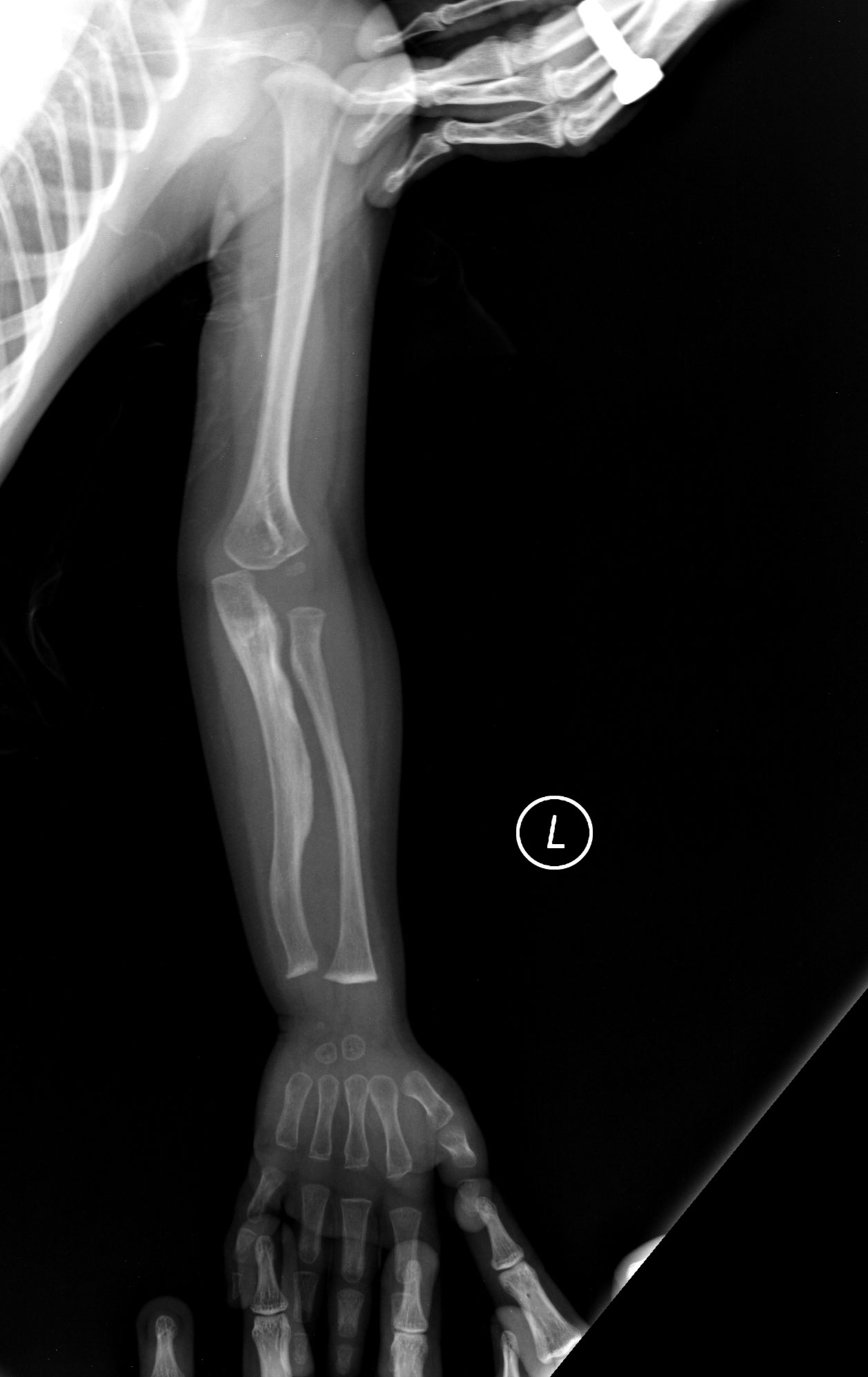
- kergesti tekkivad luumurrud, mõnikord isegi väikse traumaga;
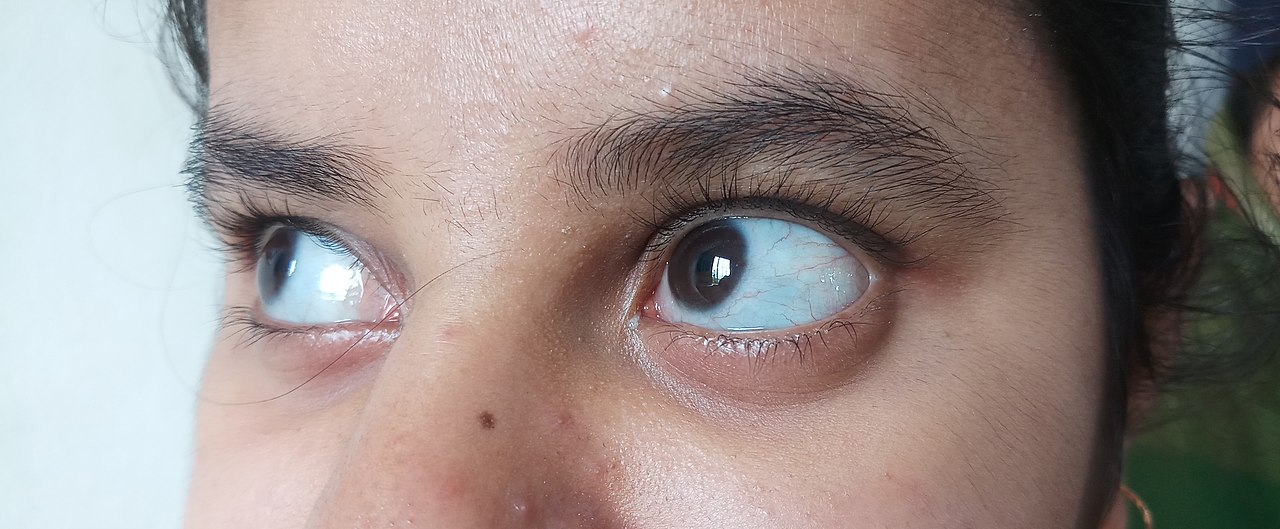
- sinakat vagy hallikat tooni silmavalged (sinised sklerad), mis tuleneb õhukesest sidekoest silma all;
- haprad või ebakorrapärased hambad (dentinogenesis imperfecta), milles hambad võivad olla haprad ja värvunud;
- kuulmislangus — paljudel täiskasvanutel võib esineda osaline kuulmislangus (ligikaudu kuni 50% juhtudest sõltuvalt tüübist);
- lühike kasv ja luude deformatsioonid (näiteks rööbas- või kumer selg — skolioos);
- liigeste painduvus või lõtvus ning lihasjõu vähenemine;
- raskematel juhtudel hingamisraskused, eriti kui rindkere on deformeerunud.
Tüübid (Sillence’i klassifikatsioon ja variatsioonid)
Osteogenesis imperfecta jagatakse kliiniliselt mitmeks tüübiks vastavalt sümptomite raskusele ja kulule. Levinuim klassifikatsioon on Sillence’i süsteem, kus peamised tüübid on:
- Tüüp I — kergeim ja kõige sagedasem vorm; sageli normaalse või vähendatud murdumissagedusega, sinised sklerad, osaline hambaprobleem ja võimalik hiline kuulmislangus;
- Tüüp II — väga raske, sageli perinataalne (sünni ümbruses) ja eluohtlik; lootel või vastsündinul võib olla mitu murrangut ja hingamisraskused, eluiga on sageli lühike;
- Tüüp III — progresseeruv deformeeriv vorm; suur hulk murranguid juba lapsepõlves, kasvupidurdumine ja märgatavad luude deformatsioonid;
- Tüüp IV — keskmise raskusega vorm, kus sümptomid jäävad tüübi I ja III vahele; sklerad võivad olla normaalse värvusega või kergelt heledad.
Tähelepanu väärib, et tegelik haiguspilt võib olla väga individuaalne ja olemas on ka teisi alamtüüpe ning harvemini esinevaid geneetilisi variatsioone.
Diagnoos
Haiguse kahtlus põhineb sageli kliinilisel pildil ja perekonna anamneesil. Diagnoosi kinnitamiseks ja täpsustamiseks kasutatakse:
- röntgeniuuringud ja luu struktuuri hindamine;
- luutiheduse mõõtmine (DXA), et hinnata luude mineraalainesisaldust;
- genetiline testimine, mis otsib mutatsioone COL1A1, COL1A2 ja teistes seotud geenides;
- vajadusel sidekoe või kollageeni biokeemiline analüüs.
Ravi ja hooldus
Kuigi praegu ei ole osteogenesis imperfecta täielikku paranemist tagavat ravi, on olemas palju meetmeid, mis parandavad elukvaliteeti ja vähendavad tüsistuste riski. Ravi tähendab sageli multidistsiplinaarset lähenemist:
- murrangute ja valu ravi (ortopeedia, kipsi- ja kirurgilised meetodid);
- pika metallvardaga (rodi) sisestamine luu tugevdamiseks ja deformatsioonide korrigeerimiseks rasketel juhtudel;
- bisfosfonaadid (nt pamidroonhape, alendroonhape) võivad suurendada luutihedust ja vähendada murrangute sagedust, eriti lastel — ravi määrab spetsialist;
- füsioteraapia ja taastusravi, et hoida lihasjõudu ja parandada liikumisvõimet;
- hammaste hooldus (stomatoloogiline jälgimine) ja vajadusel proteesid või restauratsioonid;
- audioloogiline jälgimine ja vajadusel kuuldeaparaadid;
- hingamisteede jälgimine ja ravi, eriti rasketel juhtudel.
Oluline on ka ohutuse õpetus — õige tõstetehnika, kodu ja kooli sobitamine ning abivahendite kasutamine murruriskide vähendamiseks.
Geneetiline nõustamine ja rasedus
Kui perekonnas on osteogenesis imperfecta või leitud konkreetne haigust põhjustav mutatsioon, tuleks kaaluda geneetilist nõustamist. On võimalik teha suunatud geneetilist testi raseduse ajal (nt loote biopsite uuring või amniotsentees) ja teatud olukordades ka preimplantatsiooniline geneetiline diagnostika (PGD). Nõustaja aitab hinnata pärandumise riske ja selgitada valikuid.
Tüsistused ja prognoos
Prognoos sõltub suuresti haiguse tüübist: kerged vormid võimaldavad sageli täisväärtuslikku elu ja normaalset eeldatavat eluiga, rasked vormid (nt tüüp II) võivad olla elupõhised või eluohtlikud varases eas. Levinud tüsistused on korduvad murrangud, luude deformatsioonid, kuulmislangus, hambaprobleemid ning võimalikul hingamispuudulikkus raskemate rindkere deformeerumiste korral.
Millal pöörduda arsti poole
- kui lapsel esineb ettearvamatult palju murranguid;
- kui peres on teada osteogenesis imperfecta ja plaanitakse rasedust;
- kui esinevad kõne- või kuulmishäired, kiirelt süvenev valu või hingamisraskused;
- iga kord, kui on küsimusi õigetest ravivõtetest või vajadusest multidistsiplinaarseks jälgimiseks.
Kokkuvõte: Osteogenesis imperfecta on erineva raskusastmega geneetiline haigus, mille puhul luud on hapramad kollageeni häire tõttu. Kuigi täielikku ravi veel ei ole, võimaldab tänapäevane diagnostika ja mitmekülgne ravi vähendada murrangute arvu, parandada funktsiooni ja elukvaliteeti. Geneetiline nõustamine on oluline peredele, kus haigus esineb.
Sümptomid
OI vähem tõsised sümptomid võivad olla järgmised:
- kergesti murdunud luud
- lahtised ühendused
- madal lihastoonus
- sinine, lilla või hall värvus silmade tavaliselt valges osas.
- näo kolmnurkne kuju
- kalduvus skolioosi tekkeks
- haprad hambad
OI-l on palju muid tõsiseid ja surmaga lõppevaid sümptomeid, sealhulgas hingamisprobleemid ja luumuutused.
Demograafia
OI esineb võrdselt nii meestel kui ka naistel ning võib mõjutada kõiki rahvusrühmi. OI tekib emakas ja ravi ei ole olemas. Tavaliselt avastatakse see, kui keegi murrab palju luid; neil võidakse teha DNA-test OI tuvastamiseks. Seda esineb 1 juhul 20 000-st sünnist.
Küsimused ja vastused
K: Mis on osteogenesis imperfecta?
V: Osteogenesis imperfecta on geneetiline häire, mida tavaliselt nimetatakse hapraks luuhaiguseks. See nõrgestab või hävitab kollageenivarda, mis annab luu tugevuse, ja põhjustab luude suurema murdumisohu.
K: Kuidas pärandub osteogenesis imperfecta?
V: Osteogenesis imperfecta on tavaliselt autosoomselt domineeriv haigus, mis tähendab, et inimene võib seda haigust saada, kui ainult ühel tema vanemal on ebanormaalne geen.
K: Kes tuvastas esimesena osteogenesis imperfecta?
V: Vrolik tuvastas osteogenesis imperfecta esimesena 1849. aastal.
K: Kas osteogenesis imperfecta on ravitav?
V: Kahjuks ei ole osteogenesis imperfecta ravi.
K: Millised on osteogenesis imperfecta neli tüüpi?
V: Osteogenesis imperfecta neli tüüpi on esimene, teine, kolmas ja neljas tüüp.
K: Millised on kolmanda tüübi osteogenesis imperfecta sümptomid?
V: Kolmandat tüüpi osteogenesis imperfecta'ga inimestel võib olla enne puberteeti rohkem kui 100 luumurdu. Nende silmad omandavad sageli lilla, sinise või halli varjundi ja selle juhtumi puhul on inimestel sageli ka kuulmislangus.
K: Kui levinud on kuulmislangus osteogenees imperfecta'ga inimestel?
V: 50% inimestest, kellel on osteogenesis imperfecta, on täiskasvanuks saades kuulmislangus.
Seotud artiklid
Autor
AlegsaOnline.com Osteogenesis imperfecta (hapra luuhaigus): põhjused, sümptomid ja tüübid Leandro Alegsa
URL: https://et.alegsaonline.com/art/73422
Allikad
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"