Pulmonaalne hüpertensioon (kopsuhüpertensioon) — põhjused, sümptomid
Pulmonaalne (kopsu) hüpertensioon — põhjused, sümptomid ja ravi. Kuidas ära tunda hingamisraskusi, pearinglust ja väsimust ning millal pöörduda arsti poole.
Pulmonaalne hüpertensioon ehk PH on seisund, mille puhul on kopsudes kõrge vererõhk. See seisund raskendab hingamist. Mõned inimesed, kellel on see seisund, vajavad lisahapnikku. See seisund võib põhjustada ka pearinglust ja kerget väsimust. Mõned selle haigusega inimesed minestavad kergesti. Sümptomid süvenevad treeningu või raske töö ajal. Kopsuhüpertensioon on tõsine seisund ja see võib lõppeda surmaga. See seisund raskendab südame verepumpamist. Kuna süda peab töötama raskemini, võib ta ka haigestuda. Mõned inimesed, kes on väga haiged, võivad eluks vajada kopsusiirdamist või südame-kopsusiirdamist. Pulmonaalse hüpertensiooni täisnimetus on pulmonaalne arteriaalne hüpertensioon, kuigi enamik inimesi nimetab seda pah, ph või pha.
Pildigalerii
9 Pildid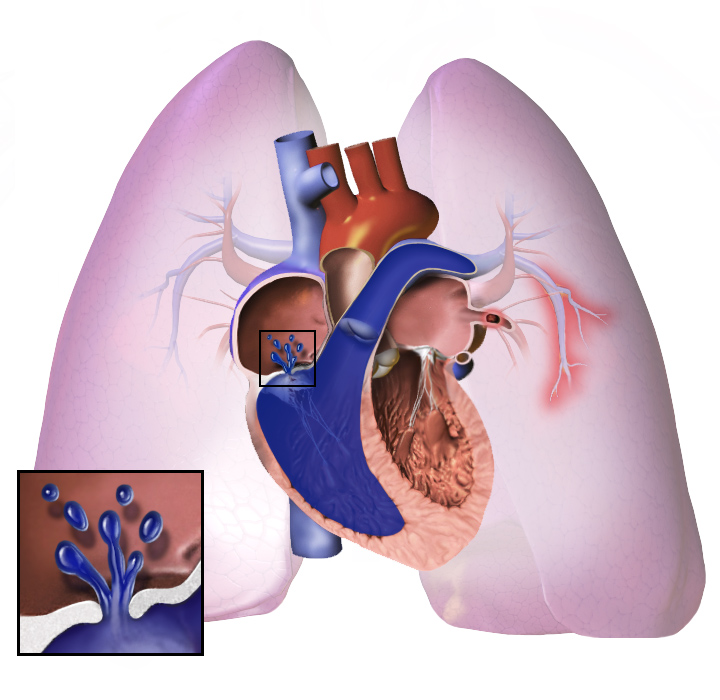






Põhjused ja tüüpide jaotus
Pulmonaalne hüpertensioon ei ole üksainus haigus, vaid mitme erineva põhjuste rühm. Arvestatakse tavaliselt järgmisi gruppe:
- Pulmonaalne arteriaalne hüpertensioon (PAH) — võib olla idiopaatiline, pärilik või seotud ravimite/toxiinide, autoimmuunhaiguste (näiteks sklerodermia), HIV-infektsiooni või kaasasündinud südamehaigustega.
- PH vasaku südamehaiguse korral — tekib siis, kui vasakpoolse südamehaiguse (nt südameklapihaigused või pärgarterite puudulikkus) tõttu tõuseb vererõhk kopsuarterites.
- PH kopsuhaiguste ja hüpoksia tõttu — kroonilised obstruktiivsed kopsuhaigused (KOPD), uneapnoe ja muud kopsupõhjused, mis vähendavad hapniku taset ja tõstavad vererõhku kopsudes.
- Krooniline trombemboolne pulmonaalne hüpertensioon (CTEPH) — korduvad trombid või kopsuarteri osa püsiv ummistus, mis tõstab vererõhku.
- Segatüübid ja mitmetahulised põhjused — mõnel inimesel on mitu samaaegset riskitegurit või põhjuseid.
Sümptomid ja märgid
Varases staadiumis võivad sümptomid olla kerged või kergesti eksitavad. Levinumad nähud ja sümptomid:
- õhupuudus, algselt pingutuse ajal, hiljem ka puhkeolekus;
- väsimus ja nõrkus;
- pearinglus või minestamine (sünkoop);
- rinnavalu või survetunne;
- jalaluu- või kõhuvalu ning tursed (vedeliku kogunemine) — märgid parema vatsakese koormusest;
- kiire või ebareeglipärane südametegevus (palpitatsioonid);
- sinakas nahavärv (tsüanoos) raskematel juhtudel.
Arst võib kuulda kontrollimisel tugevat kopsuarteri klapi sulgumise heli (tugev P2), parema vatsakese lööke või vedeliku kogunemise märke.
Kuidas diagnoositakse
Diagnoos nõuab kombineeritud uuringuid:
- esialgne hindamine ja anamnees; elektro- ja füüsikaline uuring;
- kõhu- ja rindkere röntgen, mis võib näidata kopsuteede või südame suuruse muutusi;
- EKG, mis võib näidata parema südame koormust;
- ehhokardiograafia — mitteinvasiivne uuring, mis annab esmase hinnangu kopsuarteri rõhule ja parema vatsakese funktsioonile;
- täpne diagnostiline kuldstandard on parema südame kateeteriseerimine (right heart catheterization), mis mõõdab kopsuarteri rõhku otseselt;
- täiendavad uuringud: kopsufunktsiooni testid, CT- või MRI-uuringud, V/Q-skaneerimine (eriti CTEPH kahtluse korral), vereanalüüsid (nt NT-proBNP) ja liigse trombide uurimine.
Ravi ja juhtimine
Ravi sõltub PH tüübist ja raskusastmest. Eesmärgid on sümptomite leevendamine, haiguse progresseerumise aeglustamine ja elukvaliteedi parandamine.
- Elustiili ja toetav ravi: hapnikravi vajadusel, diureetikumid vedeliku vähendamiseks, soolapiirang, korrapärane arsti jälgimine ja vaktsineerimine hingamisteede infektsioonide vastu.
- Spetsiifilised medikamentoossed ravimid: endoteliini retseptori antagonistid (nt bosentan, ambrisentan), fosfodiesteraasi-5 inhibiitorid (nt sildenafil, tadalafil), prostatsükliin-analoogid ja prostatsükliini retseptori agonistid (epoprostenool, treprostinil, selexipag), ning rüülofüllaatorid nagu riociguat teatud juhtudel.
- Antikoagulandid võivad olla vajalikud eriti kroonilise trombemboolse PH korral.
- Vasoreaktsiooni korral (teatud patsiendid täheldavad veresoonte lühiajalist avanemist) võivad mõned patsiendid kasu saada kõrgete annuste kaltsiumiantagonistidest.
- Palliatiivne ja kirurgiline sekkumine: raske ja medikamentoosselt ravimatu CTEPH võib mõnikord sobida kopsuarteri trombektoomia (kirurgiline eemaldamine). Kaugemas staadiumis võib kaaluda kopsusiirdamist või südame-kopsusiirdamist.
Eluiga ja prognoos
Prognoos sõltub PH põhjusest, avastamise faasist ja ravi alustamisest. Tänapäeval on olemas palju efektiivseid ravimeetodeid, mis on parandanud prognoosi võrreldes varasemate aastakümnetega, kuid haigus võib siiski olla eluohtlik, kui seda ei ravita adekvaatselt. Regulaarne jälgimine spetsialisti poolt ja varajane ravi alustamine on olulised tulemuse parandamiseks.
Kellele pöörduda ja millal otsida abi
- Pöörduge arstile, kui teil on selgitamata õhupuudus, korduvad minestamised, püsiv väsimus, rindkerevalu või nähtavad tursed.
- Kui teil on teadaolev riskifaktor (nt sidekoehaigus, HIV, kopsuhaigus või varasemad kopsuembooliad), rääkige arstiga PH-st ja võimalusest saata teid eriarsti juurde.
- Kiirabi tuleks kutsuda ägeda raskendava hingamisraskuse, tugeva rindkerevalu või pikaajalise teadvusekaotuse korral.
Ennetus ja päevakontroll
Mõned PH vormid ei ole täielikult ärahoitavad, kuid haiguse riskitegureid saab mõnevõrra vähendada: suitsetamisest loobumine, krooniliste kopsuhaiguste hea ravi, tromboosi riskifaktorite vähendamine ja arsti juhendamine ravimite osas, mis võivad PH-i süvendada. Rääkige arstiga raseduse plaanimisest — rasedus võib PH puhul olla eriti ohtlik ning nõuab hoolikat planeerimist ja erikohtlemist.
Kui vajate konkreetsemat teavet diagnoosi, uurimiste või ravi kohta, konsulteerige perearsti või pulmonaalspetsialistiga. Õigeaegne diagnostika ja sobiv ravi parandavad oluliselt elukvaliteeti ja prognoosi.
Märgid ja sümptomid
Kopsuhüpertensiooniga inimestel on hingamisraskused. Samuti väsivad nad kergesti. Mõned neist langevad ka kergesti minestusse. Neil võib esineda valu rinnus. Mõnedel patsientidel on jalgade ja pahkluude turse. Need sümptomid süvenevad treeningu või raske töö ajal.
Kuna paljud haigused võivad raskendada hingamist, peab arst tundma patsiendi tausta. See aitab arstil patsienti ravida, isegi kui patsiendil on mõni muu haigus. Arst teeb ka mitmeid teste. Pulmonaalne hüpertensioon muudab südame kõla. Üks test on vererõhu mõõtmine kopsuarteris, veresoones, mis läheb südamest kopsudesse.
Põhjuse kindlakstegemiseks teeb arst üldjuhul põhjaliku anamneesi. Selleks, et teha kindlaks, kas haigus võib olla perekondlik, võetakse üksikasjalik perekonna anamnees. Oluliseks peetakse kokkupuudet kokaiini, metamfetamiini, alkoholiga, mis põhjustab tsirroosi, ja suitsetamist, mis põhjustab emfüseemi. Füüsilisel läbivaatusel otsitakse pulmonaalse hüpertensiooni tüüpilisi tunnuseid, sealhulgas valju P2 (pulmonaalklapi sulgemisheli), (para)sternaalset kerkimist, jugulaarveeni paisumist, pedaalödeemi, astsiiti, hepatojugulaarse refluksi, klubilisust jmt.
Mis läheb kehaga valesti
Kopsuhüpertensiooni korral muutuvad kopsude veresooned liiga kitsaks. Vererõhk kopsudes muutub kõrgeks. Süda töötab väga kõvasti, et pumbata verd läbi kitsaste veresoonte. Hiljem muutuvad kopsude veresooned kõvaks ja paksuks. Süda peab töötama raskemini.
Süda võib töötada nii kõvasti, et see muutub haigeks. Seda nimetatakse südamepuudulikkuseks. Haige süda ei suuda verd hästi pumbata. Kopsudesse jõuab vähem verd, mistõttu veri saab vähem hapnikku. Seetõttu on raske hingata. See süveneb, kui teeme trenni või töötame kõvasti.
Põhjustab
Kõige sagedasem kopsuhüpertensiooni põhjus on vasakpoolne südamepuudulikkus. See põhjustab pulmonaalveeni hüpertensiooni. See põhjustab kopsuödeemi ehk vedeliku kogunemist kopsudesse.
Paljud haigused võivad põhjustada pulmonaalset arteriaalset hüpertensiooni (PAH).
- Kopsuhaigused, mille tõttu veres on vähem hapnikku, näiteks:
· krooniline obstruktiivne kopsuhaigus ehk KOK
· interstitsiaalne kopsuhaigus
· Pickwicki sündroom
- immuunsüsteemi probleemid, näiteks:
· AIDS
· skleroderma
· muud autoimmuunhaigused
- maksaprobleemid
· tsirroos
· portaalne hüpertensioon
- muud põhjused
· uneapnoe
· kaalulangetuspillide, nagu Fen-Phen, Aminorex, fenfluramiin (Pondimin) ja fentermiin, võtmine.
· sirprakuline haigus,
· kaasasündinud südamehaigus
· kilpnäärme haigused,
· narkootikumide, näiteks kokaiini tarvitamine
· võimalik, et inimese herpesviirus 8
Kui inimesel on kopsuhüpertensioon ilma muu põhjuseta, nimetatakse seda idiopaatiliseks pulmonaalseks arteriaalseks hüpertensiooniks või IPAH-ks.
Kui haigus esineb perekonnas, nimetatakse seda haigust perekondlikuks pulmonaalseks arteriaalseks hüpertensiooniks (FPAH). IPAH-d ja FPAH-d peetakse nüüd geneetilisteks häireteks, mis on seotud mutatsioonidega geenis BMPR2, mis kodeerib luu morfogeneetiliste valkude retseptorit, ning geenis 5-HT(2B), mis kodeerib serotoniini retseptorit.
Meditsiinis on pulmonaalhüpertensioon (PH) vererõhu tõus kopsuarteris või kopsuveresoonkonnas, mis põhjustab õhupuudust, pearinglust, minestust ja muid sümptomeid, mis kõik süvenevad pingutuse korral. Sõltuvalt põhjusest võib kopsuhüpertensioon olla raske haigus, millega kaasneb märgatavalt vähenenud koormustaluvus ja parempoolne südamepuudulikkus. Seda tuvastas esmakordselt dr Ernst von Romberg 1891. aastal. See võib olla üks viiest erinevast tüübist, arteriaalne, venoosne, hüpoksiline, trombembooliline või muu.
Kuigi patsientidele ja üldsusele levitatavates materjalides kasutatakse endiselt termineid primaarne pulmonaalne hüpertensioon (mis tähendab teadmata põhjusega) ja sekundaarne pulmonaalne hüpertensioon (mis on tingitud mõnest muust haigusest), on need terminid meditsiinikirjanduses suures osas kõrvale jäetud. See muutus on toimunud seetõttu, et vanem dihhotoomiline klassifikatsioon ei kajastanud patofüsioloogiat ega tulemust. See tõi kaasa ekslikud raviotsused, st ainult "primaarse" pulmonaalse hüpertensiooni ravi. See omakorda tõi kaasa terapeutilise nihilismi paljude "sekundaarse" pulmonaalse hüpertensiooniga tähistatud patsientide puhul ja võis kaasa aidata nende surmajuhtumite tekkimisele. Termin "primaarne pulmonaalne hüpertensioon" on nüüdseks asendatud terminiga "idiopaatiline pulmonaalne arteriaalne hüpertensioon". Mõisteid "primaarne" ja "sekundaarne" pulmonaalne hüpertensioon ei tohiks enam kasutada. Täiendavad üksikasjad on esitatud allpool jaotises "Klassifikatsioon".
Põhjustab
Kõige sagedasem pulmonaalse hüpertensiooni põhjus on vasaku südame puudulikkus, mis põhjustab pulmonaalveeni hüpertensiooni. Selle põhjuseks võib olla vasaku vatsakese süstoolne või diastoolne talitlushäire või klapihäire, näiteks mitraalregurgitatsioon või mitraalstenoos. Tavaliselt avaldub see kopsuödeemina.
Pulmonaalse arteriaalse hüpertensiooni (PAH) levinumad põhjused on HIV, skleroderma ja muud autoimmuunhaigused, tsirroos ja portaalhüpertensioon, sirprakuline haigus, kaasasündinud südamehaigused, kilpnäärmehaigused jt. Kaalulangetuspillide, nagu Fen-Phen, Aminorex, fenfluramiin (Pondimin) ja fentermiin, kasutamine on varem viinud PAH-i tekkimiseni.
Patogenees
Olenemata algsest põhjusest on kopsuhüpertensioon seotud kopsudega seotud ja neis asuvate veresoonte ahenemisega. See muudab südame jaoks raskemaks vere pumpamise läbi kopsude, nii nagu on raskem panna vesi voolama läbi kitsa toru kui läbi laia toru. Aja jooksul muutuvad kahjustatud veresooned nii jäigemaks kui ka paksemaks, suurendades veelgi vererõhku kopsudes ja kahjustades verevoolu. Lisaks põhjustab südame suurenenud töökoormus parema vatsakese pikenemist ja laienemist, mistõttu süda ei suuda enam nii hästi verd läbi kopsude pumbata, põhjustades parema südame puudulikkust. Kuna kopsude kaudu voolav veri väheneb, saab südame vasakpoolne osa vähem verd. See veri võib kanda ka vähem hapnikku kui tavaliselt. Seetõttu on südame vasakpoolsel poolel üha raskem pumbata, et anda ülejäänud kehale piisavalt hapnikku, eriti füüsilise tegevuse ajal.
Diagnoos
Kuna pulmonaalne hüpertensioon võib olla 5 peamist tüüpi, tuleb teha rida teste, et eristada pulmonaalset arteriaalset hüpertensiooni venoosse, hüpoksilise, tomboemboolilise või muu liigiga hüpertensioonist.
Füüsilise läbivaatuse käigus otsitakse kopsuhüpertensiooni tüüpilisi tunnuseid. Nende hulka kuuluvad muutunud südamehelid, nagu laialt lõhestatud S2 või teine südameheli, valju P2 või pulmonaalklapi sulgemisheli (osa teisest südamehelist), (para)sternaalne tõus, võimalik S3 või kolmas südameheli ja pulmonaalne regurgitatsioon. Muude tunnuste hulka kuuluvad jugulaarveenide paisumine (jugulaarveenide laienemine), perifeerne turse (pahkluude ja jalgade turse), astsiit (vedeliku kogunemisest tingitud kõhupuhitus), hepatojugulaarne refluks ja kopsupõletik.
Pulmonaalse hüpertensiooni olemasolu kinnitamiseks ja muude võimalike diagnooside välistamiseks on vaja täiendavaid protseduure. Need hõlmavad tavaliselt kopsufunktsiooni teste, vereanalüüse, elektrokardiograafiat (EKG), arteriaalse vere gaaside mõõtmist, röntgenülesvõtteid rindkerest (millele järgneb kõrge resolutsiooniga kompuutertomograafia, kui kahtlustatakse interstitsiaalset kopsuhaigust) ja ventilatsiooni-perfusiooni või V/Q-skaneerimist, et välistada krooniline trombembooliline kopsuhüpertensioon. Kopsu biopsia ei ole tavaliselt näidustatud, välja arvatud juhul, kui kahtlustatakse, et pulmonaalhüpertensioon on tingitud interstitsiaalsest kopsuhaigusest. Kuid kopsude biopsiaga kaasneb verejooksu oht kõrge intrapulmonaalse vererõhu tõttu. Kliinilist paranemist mõõdetakse sageli "kuue minuti kõndimistesti" abil, st kui kaugele patsient suudab kuue minutiga kõndida. Selle mõõtmise stabiilsus ja paranemine on seotud parema elulemusega.
Kuigi kopsuarteri rõhku saab hinnata ehhokardiograafia alusel, on kõige kindlam mõõtmine võimalik Swan-Ganz'i kateetriga võetud rõhu proovide abil. PAOPi ja PVRi ei saa otse ehhokardiograafia abil mõõta. Seetõttu on PAH diagnoosimiseks vaja südame kateeterdamist. Swan-Gansi kateetriga saab mõõta ka südame väljavoolu, mis on haiguse raskusastme mõõtmisel palju olulisem kui kopsuarteri rõhk.
Normaalne kopsuarteri rõhk merepinna kõrgusel elaval inimesel on keskmiselt 12-16 mm Hg (1600-2100 Pa). Kindel pulmonaalhüpertensioon on olemas, kui keskmine rõhk puhkeseisundis ületab 25 mm Hg (3300 Pa). Kui kopsuarteri keskmine rõhk tõuseb koormuse korral üle 30 mm Hg (4000 Pa), loetakse seda samuti pulmonaalhüpertensiooniks.
PAH diagnoosimiseks on vajalik pulmonaalse hüpertensiooni olemasolu koos kahe muu seisundiga. Kopsuarteri oklusioonirõhk (PAOP või PCWP) peab olema väiksem kui 15 mm Hg (2000 Pa) ja kopsuveresoonte vastupanu (PVR) peab olema suurem kui 3 Woodi ühikut (240 dyn-s-cm−5 või 2,4 mN-s-cm−5 ).
Klassifikatsioon
Praegune klassifikatsioon
2003. aastal kutsuti Veneetsias kokku 3. ülemaailmne kopsuarteri hüpertensiooni sümpoosion, et muuta klassifikatsiooni vastavalt uuele arusaamisele haiguse mehhanismidest. Selle rühma poolt välja töötatud läbivaadatud süsteem on praegune raamistik pulmonaalse hüpertensiooni mõistmiseks.
Süsteem sisaldab mitmeid parandusi võrreldes endise, 1998. aasta Eviani klassifikatsioonisüsteemiga. Riskifaktorite kirjeldusi on ajakohastatud ja kaasasündinud süsteemi-pulmonaalsete šunte klassifitseeritud. Soovitati luua uus geneetiliste faktorite klassifikatsioon, kuid seda ei rakendatud, sest olemasolevaid andmeid peeti ebapiisavaks.
Veneetsia 2003. aasta läbivaadatud klassifikatsioonisüsteemi võib kokku võtta järgmiselt:
- WHO I rühm - pulmonaalne arteriaalne hüpertensioon (PAH)
- WHO II rühm - vasaku südame haigusega seotud pulmonaalne hüpertensioon
- WHO III rühm - kopsuhaiguste ja/või hüpokseemiaga seotud pulmonaalne hüpertensioon
- WHO IV rühm - kroonilisest tromboosist ja/või emboossest haigusest tingitud pulmonaalne hüpertensioon
- WHO V rühm - Mitmesugused
Eelmine terminoloogia
Varem kasutati haiguse liigitamiseks mõisteid primaarne ja sekundaarne pulmonaalne hüpertensioon (PPH ja SPH). See tõi kaasa eelduse, et ravida tuleks ainult esmast haigust ja teiseseid vorme tuleks ignoreerida, et ravida ainult põhihaigust. Tegelikult on kõik pulmonaalse arteriaalse hüpertensiooni vormid ravitavad. Kahjuks püsib see liigitussüsteem paljude arstide teadvuses ja viib tõenäoliselt selleni, et paljudele patsientidele ei anta ravi. Selline nihilistlik lähenemine pulmonaalsele arteriaalsele hüpertensioonile võib kaasa aidata ka aladiagnoosimisele. Hinnanguliselt on USAs umbes 100 000 PAHiga patsienti, kuid ainult 15-20 000 on diagnoositud. Paljudel teistel on diagnoositud vääralt KOK, astma või südamepuudulikkus.
Mõiste primaarne pulmonaalne hüpertensioon (PPH) on nüüdseks suures osas meditsiinikirjanduses asendatud idiopaatilise pulmonaalse arteriaalse hüpertensiooniga (IPAH). Mõned arstid kasutavad siiski jätkuvalt vanemat klassifikatsiooni ebasobivalt.
Epidemioloogia
IPAH on haruldane haigus, mille esinemissagedus on umbes 2-3 miljonit inimest aastas ja levimus umbes 15 miljonit inimest. Naistel esineb IPAH peaaegu kolm korda sagedamini kui meestel.
Muud PAHi vormid on palju sagedamini esinevad. Skleroderma korral on esinemissagedus hinnanguliselt 6-60% kõigist patsientidest, reumatoidartriidi korral kuni 21%, süsteemse erütematoosse luupuse korral 4-14%, portaalhüpertensiooni korral 2-5%, HIVi korral umbes 0,5% ja sirprakulise haiguse korral 20-40%.
Dieetpillid, nagu Fen-Phen, tekitasid aastas 25-50 juhtumit miljoni inimese kohta aastas.
Treatment
Ravi sõltub sellest, kas PH on arteriaalne, venoosne, hüpoksiline, trombembooliline või muu. Kuna pulmonaalveeniline hüpertensioon on sünonüümne kongestiivse südamepuudulikkusega, on raviks vasaku vatsakese funktsiooni optimeerimine diureetikumide, beetablokaatorite, AKE-inhibiitorite jne abil või mitraalklapi või aordiklapi parandamine/asendamine.
PAH puhul peetakse tavapäraseks raviks elustiili muutusi, digoksiini, diureetikume, suukaudseid antikoagulante ja hapnikravi, kuid nende kasulikkust ei ole kunagi tõestatud randomiseeritud, prospektiivsel viisil.
Kõrge annusega kaltsiumikanali blokaatorid on kasulikud ainult 5% IPAH patsientidest, kes on Swan-Gansi kateetri abil vasoreaktiivsed. Kahjuks on kaltsiumikanali blokaatoreid suures osas vääralt kasutatud, kuna neid on määratud paljudele mittevasoreaktiivse PAH-ga patsientidele, mis põhjustab liigset haigestumust ja suremust.
Vasoaktiivsed ained
Kopsuarteri silelihasrakkude ebanormaalses proliferatsioonis ja kontraktsioonis on kopsuarteri arteriaalse hüpertensiooniga patsientidel osalenud kolm peamist teed. Need teed vastavad selle haiguse puhul olulistele terapeutilistele sihtmärkidele ja mängivad rolli selle määramisel, milliseid kolmest ravimiklassist - endoteliini retseptori antagonistid, fosfodiesteraasi tüüp 5 inhibiitorid ja prostatsükliiniderivaadid - kasutatakse.
Prostatsükliini (prostaglandiin I2 ) peetakse tavaliselt kõige tõhusamaks PAHi raviks. Epoprostenooli (sünteetiline prostatsükliin, mida turustatakse nime all Flolan®) manustatakse pideva infusioonina, mis nõuab poolpüsivat tsentraalset veenikatetrit. See manustamissüsteem võib põhjustada sepsist ja tromboosi. Flolan® on ebastabiilne ja seetõttu tuleb seda manustamise ajal hoida jääl. Kuna selle poolväärtusaeg on 3-5 minutit, peab infusioon olema pidev (24/7) ja selle katkestamine võib lõppeda surmaga. Seetõttu on välja töötatud teisi prostanoide. Treprostiniili (Remodulin®) võib manustada intravenoosselt või subkutaanselt, kuid subkutaanne vorm võib olla väga valulik. Iloprosti (Ilomedin®) kasutatakse Euroopas samuti intravenoosselt ja selle poolväärtusaeg on pikem. Iloprost (mida turustatakse kui Ventavis®) on ainus USAs ja Euroopas kasutamiseks heakskiidetud inhaleeritav prostatsükliini vorm. Selle manustamisviisi eeliseks on selektiivne ladestumine kopsudes, millel on vähem süsteemseid kõrvaltoimeid.
2001. aastal kiideti heaks kahekordne (ETA ja ETB ) endoteliiniretseptori antagonist bosentaan (turustatakse nime Tracleer® all). Kaks selektiivset endoteliiniretseptori antagonisti (ainult ETA ) on heakskiitmise lõppjärgus: sitaxsentaan ja ambrisentaan. Sildenafiil, cGMP-spetsiifilise fosfodiesteraasi 5. tüübi (PDE5) selektiivne inhibiitor, kiideti PAHi raviks heaks 2005. aastal. Seda turustatakse PAHi puhul Revatio® nime all. Tadalafiili (mida praegu turustatakse erektsioonihäirete puhul Cialis® nime all) uuritakse praegu III faasi uuringutes. Vasoaktiivne soolepeptiid inhalatsiooni teel peaks sisenema kliinilistesse uuringutesse PAHi raviks 2007. aastal. PRX-08066 on serotoniini antagonist, mida praegu töötatakse välja hüpoksilise pulmonaalse hüpertensiooni raviks.
Kirurgiline
Atrial septostoomia on kirurgiline protseduur, millega luuakse ühendus parema ja vasaku eesoja vahel. See vähendab rõhku südame paremal poolel, kuid selle hinnaga väheneb hapniku sisaldus veres (hüpoksia). Seda on kõige parem teha kogenud keskustes. Kopsutransplantatsioon ravib kopsuarteriüpertensiooni, kuid jätab patsiendile tüsistused ja umbes 5-aastase elulemuse.
Pulmonaalne trombendarterektoomia (PTE) on kirurgiline protseduur, mida kasutatakse kroonilise trombemboolilise pulmonaalse hüpertensiooni korral. See on organiseeritud trombi (trombi) kirurgiline eemaldamine koos kopsuarteri limaskestaga; see on suur ja väga raske protseduur, mida praegu tehakse vaid vähestes valitud keskustes. Juhtumite seeriad näitavad enamiku patsientide puhul märkimisväärset edu.
Kopsuhüpertensiooni hüpoksilise ja mitmesuguste variatsioonide ravi ei ole kindlaks tehtud. Siiski on praegu käimas mitme toimeaine uuringud, kuhu võetakse patsiente. Paljud arstid ravivad neid haigusi samade ravimitega nagu PAH puhul, kuni paremad võimalused muutuvad kättesaadavaks.
Prognoos
1980. aastate NIH IPAH registri andmetel oli ravimata elulemuse mediaan 2-3 aastat alates diagnoosimisest, kusjuures surma põhjuseks oli tavaliselt parempoolse vatsakese puudulikkus (cor pulmonale). Kuigi seda näitajat tsiteeritakse laialdaselt, on see tänapäeval tõenäoliselt ebaoluline. Tulemused on viimase kahe aastakümne jooksul oluliselt muutunud. See võib olla tingitud uuemast ravimitest, paremast üldisest hooldusest ja varasemast diagnoosimisest (varajane diagnoosimine). Hiljuti läbi viidud tulemusuuring nende patsientide kohta, kes alustasid ravi bosentaaniga (Tracleer®), näitas, et 86% patsientidest olid 3 aasta pärast elus. Kuna nüüd on saadaval mitu ainet, kasutatakse üha enam kombinatsioonravi. Nende ainete mõju elulemusele ei ole teada, sest paljud neist on välja töötatud alles hiljuti. Ei oleks põhjendamatu eeldada, et lähitulevikus ületab keskmine elulemus 10 aastat.
Küsimused ja vastused
K: Mis on pulmonaalne hüpertensioon ehk PH?
V: Pulmonaalne hüpertensioon ehk PH on seisund, kus kopsudes on kõrge vererõhk.
K: Millised on kopsuhüpertensiooni sümptomid?
V: Kopsuhüpertensiooni sümptomiteks on hingamisraskused, pearinglus, väsimus ja minestus.
K: Miks vajavad mõned pulmonaalse hüpertensiooniga inimesed lisahapnikku?
V: Mõned pulmonaalse hüpertensiooniga inimesed vajavad lisahapnikku, sest haiguse tõttu on neil raske hingata.
K: Millal süvenevad kopsuhüpertensiooni sümptomid?
V: Pulmonaalhüpertensiooni sümptomid süvenevad treeningu või raske töö ajal.
K: Miks on pulmonaalhüpertensioon tõsine seisund?
V: Pulmonaalne hüpertensioon on tõsine seisund, sest see raskendab südame verepumpamist ja võib lõppeda surmaga.
K: Mis on pulmonaalhüpertensiooni täisnimetus?
V: Pulmonaalse hüpertensiooni täisnimetus on pulmonaalne arteriaalne hüpertensioon, kuigi enamik inimesi nimetab seda pah, ph või pha.
K: Mida võivad mõned väga haiged inimesed, kellel on pulmonaalhüpertensioon, eluks vajada?
V: Mõned väga haiged inimesed, kellel on pulmonaalhüpertensioon, võivad eluks vajada kopsusiirdamist või südame-kopsusiirdamist.
Seotud artiklid
Autor
AlegsaOnline.com Pulmonaalne hüpertensioon (kopsuhüpertensioon) — põhjused, sümptomid Leandro Alegsa
URL: https://et.alegsaonline.com/art/80022
Allikad
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171