Mis on molekulaarorbitaal? Selgitus, omadused ja näited
Mis on molekulaarorbitaal? Selge sissejuhatus, kvantmehaaniline seletus, omadused ja praktilised näited — õppige, kuidas orbitaalid määravad molekulide struktuuri ja käitumist.
Keemias selgitab molekulaarorbitaal (või MO), mis juhtub elektronidega, kui aatomid ühinevad molekulis. MO on matemaatiline funktsioon, mis kirjeldab elektroni lainepõhist käitumist molekulis. Keemikud kasutavad selliseid funktsioone keemiliste ja füüsikaliste omaduste ennustamiseks või seletamiseks. Näiteks võivad funktsioonid öelda, kui tõenäoline on elektroni leidmine mõnes konkreetses piirkondas.
Keemikud koostavad molekulaarorbitaalide matemaatilisi mudeleid tavaliselt aatomiorbitaalide kombineerimise teel. Kasutada võib ka molekuli iga aatomi hübriidorbitaale või muid aatomirühmade molekulaarorbitaale. Arvutid võivad töötada nende funktsioonidega. Molekulaarorbitaalid võimaldavad keemikutel rakendada molekulide uurimisel kvantmehaanikat. MO-d vastavad küsimustele, kuidas aatomid molekulides koos püsivad. Erinevad ümarad kujud orbitaaldiagrammil näitavad, kus aatomi elektronid kõige tõenäolisemalt paiknevad.
Pildigalerii
5 Pildid
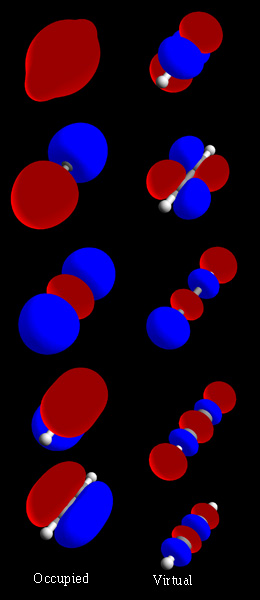



Mida molekulaarorbitaal tegelikult tähendab?
Molekulaarorbitaal on lainefunktsioon (ψ), mis levib tavaliselt üle kogu molekuli — see tähendab, et üks MO ei kuulu ainult ühe aatomini, vaid võib olla hajunud mitme aatomi vahel. Elektroni leidmise tõenäosus selles orbitaalil on proportsionaalne psi ruuduga (|ψ|^2), mida sageli kuvatakse elektronidensiteedina.
Kuidas MO tekib — siduvad ja vastupidavad orbitaalid
Molekulaarorbitaalid tekivad aatomiorbitaalide kombineerimisel. Kui kahe aatomiorbitaali lainelained kokku langevad, võivad nad interferentsi teel omavahel tugevdada või nõrgestada:
- Kui fazid kattuvad konstruktivselt, tekib siduv (bonding) orbitaal, mille energia on väiksem kui lähteaatomiorbitaalide energia — see soodustab sideme tekkimist.
- Kui fazid kattuvad destruktivselt, tekib antisding (antibonding) orbitaal (märgitud sageli tähega *), millel on sõlmpinnad (nodes) ja mille energia on kõrgem — täidetud antibonding-orbitaalid nõrgestavad sidet.
Olulised mõisted ja omadused
- HOMO ja LUMO: kõrgeima energia täidetud orbitaali nimetatakse HOMO-ks (Highest Occupied Molecular Orbital) ja madalaima energia täitmata orbitaali LUMO-ks (Lowest Unoccupied Molecular Orbital). Nende vahe määrab sageli molekuli keemilise reaktiivsuse ja spektrilised omadused.
- Orbitaalitüüpide nimed: sigma (σ) ja pi (π) orbitaalid kirjeldavad sümmeetriat ümber sidetav telje: σ-orbitaalid on sümmeetrilised selle telje ümber, π-orbitaalid tekivad külgmise kattumise tulemusena ja on energiliselt tavaliselt kõrgemad kui vastavad σ-orbitaalid sama peamise tasonumbri piires.
- Sideme järjekord: lihtne hinnang sideme tugevusele: bond order = (nb − na)/2, kus nb on siduvate elektronide arv ja na antibonding-elektronide arv. Näiteks H2-l on bond order = (2 − 0)/2 = 1.
- Orbitaalide nodid: antibonding-orbitaalidel esineb sõlmpindu — piirkondi, kus psi = 0 ja kus elektronile on väga väike tõenäosus sattuda.
Lihtsad näited
- H2: kaks H aatomit kombineerivad 1s orbitaalid ja moodustavad σ1s (siduv) ning σ*1s (antibonding) orbitaalid. Kaks elektroni täidavad siduva orbitaali → stabiilne ühe sidemega molekul.
- O2: hapniku molekulil on MO-ehitus, kus kaks elektronikihti täidavad π* (antibonding) orbitaalid paaritult — see selgitab O2 paramagnetilisust (kaks paaritut elektroni) ja bond order = 2.
- CO: süsiniku-monooksiidil on MO-de ja elektroni jaotuse tõttu eriline dipool ja tugev kolmikside; molekulaarorbitaalide analüüs aitab selgitada selle kiiruse ja ligipääsetavust reaktsioonidel.
Molekulaarorbitaalide moodustamise meetodid
Keemikud ja teoreetikud kasutavad erinevaid meetodeid MO-de arvutamiseks:
- LCAO (Linear Combination of Atomic Orbitals): lihtne ja intuitiivne meetod, kus MO-d kirjutatakse aatomiorbitaalide lineaarse kombinatsioonina.
- Ab initio ja tiheduse funktsionaali (DFT) meetodid: arvutuslikult täpsemad lähenemised, mida kasutatakse keerukamate molekulide korral, et ennustada MO-de energiaid ja kujusid.
Miks MO on tähtis — praktilised rakendused
- Selgitab ja ennustab molekulide magnetilisi omadusi (paramagnetism/diamagnetism).
- HOMOs ja LUMOs analüüs aitab ennustada keemilist reaktiivsust, näiteks kus elektrofiil või nukleofiil reageerib.
- Molekulide optilised omadused (imendumine, spektroskoopia) on tihedalt seotud elektronide üleminekute vahel MO-de vahel.
- Keemiliste sidemete tugevus, lõikepunktide arv ja polaarsus on mõistetavad MO-de täitumise kaudu.
Kokkuvõte
Molekulaarorbitaalid on kvantmehaanilised lainefunktsioonid, mis kirjeldavad elektronide jaotust molekulides. Need tekivad aatomiorbitaalide kombineerimisel ning jagunevad siduvateks ja antibonding-orbitaalideks. MO-diagrammid, HOMO/LUMO-kujundid ja elektronide täitumine annavad võimsad tööriistad molekulide omaduste, reaktsioonikäitumise ja optiliste fenomenide mõistmiseks ja ennustamiseks.

Ajalugu
Sõna orbitaal kasutas inglise keeles esmakordselt Robert S. Mulliken. Saksa füüsik Erwin Schrödinger kirjutas MO-dest juba varem. Schrödinger nimetas neid Eigenfunktioniks.
Füüsik Max Born kirjeldas 1926. aastal molekulaarorbitaalide teooria. Tänapäeval tuntakse seda Borni reeglina ja see on osa kvantmehaanika Kopenhaageni tõlgendusest. Kui see teooria algselt välja pakuti, ei olnud see kooskõlas Niels Bohri aatomimudeliga. Bohri mudel kirjeldas elektronide "tiirlemist" tuuma ümber, kuna nad liikusid ringiratastena. Borni mudel sai siiski lõpuks populaarsust, sest see suutis kirjeldada elektronide asukohti molekulides ja seletada mitmeid seni seletamatuid keemilisi reaktsioone.
Ülevaade
Aatomi orbitaalid ennustavad elektroni asukohta aatomis. Molekulaarorbitaalid tekivad aatomiorbitaalide ühendamisel. Molekulaarorbitaal võib anda teavet molekuli elektronide konfiguratsiooni kohta. Elektroni konfiguratsioon on ühe (või ühe paari) elektroni(de) kõige tõenäolisem asukoht ja energia. Enamasti esitatakse MO aatomiorbitaalide lineaarkombinatsioonina (LCAO-MO meetod), eriti ligikaudsel kasutamisel. See tähendab, et keemikud eeldavad, et tõenäosus, et elektron asub molekuli mis tahes punktis, on elektroni seal olemise tõenäosuste summa, mis põhineb üksikutel aatomiorbitaalidel. LCAO-MO on lihtne mudel molekulide sidemete kohta ja on oluline molekuliorbitaalide teooria uurimisel.
Teoreetilised keemikud kasutavad arvutit, et arvutada erinevate molekulide MO-d (nii reaalsed kui ka kujuteldavad). Arvuti võib joonistada "pilve" graafikuid, mis näitavad, kui tõenäoliselt asub elektron mis tahes piirkonnas. Arvutid võivad anda teavet ka molekuli füüsikaliste omaduste kohta. Samuti oskavad nad öelda, kui palju energiat on vaja molekuli moodustamiseks. See aitab keemikutel öelda, kas mõnda väikest molekuli saab ühendada, et moodustada suuremaid molekule.
Enamik tänapäevaseid arvutusliku keemia viise algab süsteemi MO-de arvutamisega. Iga MO elektrivälja tekitavad kõikide aatomite tuumad ja teiste elektronide mingi keskmine jaotus.
Analoogia
MO-de mõistmine on nagu ülesanne teada, kus iga töötaja on suures remondipoes (ilma poodi sisse vaatamata). Analüütik teab poes töötavate töötajate arvu ja iga töötaja osakonda. Samuti teab ta, et töötajad ei astu üksteise varvastele ja et töötajad seisavad pigem vahekäikudes kui kaubariiulitel. Töötajad lahkuvad oma osakonnast, et aidata klientidel leida kaupu teistes osakondades või kontrollida inventari. Analüütik, kes annab kõikide töötajate asukoha poes valitud hetkel ilma sisse vaatamata, on nagu keemik, kes arvutab molekuli MO-d. Nii nagu MO ei saa öelda iga elektroni täpset asukohta, ei ole ka iga töötaja täpne asukoht teada. MO, millel on nooditasand, on nagu järeldus, et töötajad kõnnivad mööda vahekäike, mitte läbi riiulite. Kuigi elektronid panustatakse konkreetsest aatomist, täidab elektron MO-d, arvestamata selle lähteatoomi. See on nagu töötaja, kes lahkub oma osakonnast, et päeva jooksul mujal poes jalutada. Seega on MO elektroni mittetäielik kirjeldus, nii nagu analüütiku arvutused nähtamatu poe kohta on mittetäielik oletus töötaja asukoha kohta.

Molekulaarorbitaalide moodustamine
Teoreetilised keemikud on leiutanud reeglid MO-de arvutamiseks. Need reeglid tulenevad kvantmehaanika mõistmisest. Kvantmehaanika aitab keemikutel kasutada seda, mida füüsika ütles elektronide kohta, et töötada välja, kuidas elektronid molekulides käituvad. Molekuliorbitaalid moodustuvad aatomiorbitaalide vahelistest "lubatud" vastastikmõjudest. (Interaktsioonid on "lubatud", kui aatomiorbitaalide sümmeetriad (mis on määratud rühmateooria põhjal) on omavahel ühilduvad). Keemikud uurivad aatomiorbitaalide vastastikmõjusid. Need vastastikmõjud tulenevad kahe aatomi orbitaali kattumisest (mõõt, mis näitab, kui hästi kaks orbitaali omavahel konstruktiivselt suhtlevad). Kattuvus on oluline, kui aatomiorbitaalid on energeetiliselt lähedased. Lõpuks peab molekuli MO-de arv olema võrdne molekuli moodustamiseks kokku viidavate aatomite aatomiorbitaalide arvuga.
Kvalitatiivne lähenemine
Keemikud peavad molekulide struktuuri arutamiseks mõistma MOde geomeetriat. LCMO (Linear combination of atomic orbitals molecular orbital) meetod annab MO-de ligikaudse, kuid hea kirjelduse. Selle meetodi puhul väljendatakse molekuliorbitaalid kõikide molekuli aatomi aatomiorbitaalide lineaarkombinatsioonidena.
Aatomiorbitaalide lineaarkombinatsioonid (LCAO)
Molekulaarorbitaalide kasutamist tutvustasid esimest korda Friedrich Hund ja Robert S. Mulliken 1927. ja 1928. aastal.
Aatomiorbitaalide lineaarkombinatsiooni ehk LCAO-lähenemist molekuliorbitaalide jaoks võttis 1929. aastal kasutusele Sir John Lennard-Jones. Tema teedrajav töö näitas, kuidas tuletada fluori ja hapniku molekulide elektrooniline struktuur kvantpõhimõtetest. See kvalitatiivne lähenemine molekuliorbitaalide teooriale on osa kaasaegse kvantkeemia algusest.
Aatomiorbitaalide lineaarkombinatsioonide (LCAO) abil saab ära arvata molekuli orbitaalid, mis tekivad molekuli aatomite omavahelisel sidumisel. Sarnaselt aatomiorbitaaliga saab ka molekuliorbitaalile konstrueerida Schrödingeri võrrandi, mis kirjeldab elektroni käitumist. Aatomi orbitaalide lineaarkombinatsioonid (aatomi lainekujude summad ja erinevused) annavad molekulaarsete Schrödingeri võrrandite ligikaudsed lahendused. Lihtsate kaheaatomiliste molekulide puhul esitatakse saadud lainefunktsioonid matemaatiliselt võrranditega
Ψ = c aψ a+ c bψ b
ja
Ψ* = c aψ a- c bψ b
kus Ψ ja Ψ* on vastavalt siduvate ja mittesiduvate molekulaarorbitaalide molekulaarlainefunktsioonid, ψa ja ψb on vastavalt aatomi a ja b aatomi aatomi lainekujutised ning ca ja c bon reguleeritavad koefitsiendid. Need koefitsiendid võivad olla positiivsed või negatiivsed, sõltuvalt üksikute aatomiorbitaalide energiatest ja sümmeetriatest. Kui kaks aatomit lähenevad teineteisele, kattuvad nende aatomiorbitaalid, tekitades suure elektrontihedusega alasid. Seega moodustuvad kahe aatomi vahel molekuliorbitaalid. Aatomeid hoiab koos elektrostaatiline tõmme positiivselt laetud tuumade ja negatiivselt laetud elektronide vahel, mis hõivavad siduvate molekuliorbitaalide orbitaalid.
Siduvad, mittesiduvad ja mittesiduvad MOd
Kui aatomi orbitaalid interakteeruvad, võib tekkida kolme tüüpi molekuliorbitaal: sidus, mittesiduv või mittesiduv.
Sidumisviisid:
- Aatomiorbitaalide vahelised sidemete vastastikmõjud on konstruktiivsed (faasisisesed) vastastikmõjud.
- Siduvate MO-de energia on madalam kui aatomi orbitaalide energia, mis neid ühendavad.
Sidumisvastased meetodid:
- Aatomiorbitaalide vahelised antisiduvad vastastikmõjud on destruktiivsed (faasivälised) vastastikmõjud.
- Antibonding MO-d on energeetiliselt kõrgemad kui aatomi orbitaalid, mis ühendavad neid.
Mittekohustuslikud MO-d:
- Mittesiduvad MO-d on tingitud sellest, et aatomiorbitaalide vahel puudub vastastikmõju, kuna puuduvad ühilduvad sümmeetriad.
- Mittesiduvatel MO-del on sama energia kui molekuli ühe aatomi aatomi orbitaalidel.
HOMO ja LUMO
Igal molekuliorbitaalil on oma energiatase. Keemikud sorteerivad MO-d energiatasemete järgi. Keemikud eeldavad, et elektronid täidavad kõigepealt madalaima energiatasemega MO-d. Näiteks kui molekulil on 15 orbitaali täitmiseks vajalikud elektronid, siis täidetakse 15 madalaima energiatasemega MO-d. Nimekirja 15. MO-d nimetatakse "kõrgeima hõivatud molekuliorbitaaliks" (HOMO) ja 16. MO-d "madalaima hõivamata molekuliorbitaaliks" (LUMO). HOMO energiataseme ja LUMO energiataseme erinevust nimetatakse ribalõheks. Bändilõhe võib mõnikord olla molekuli ergastatavuse mõõdupuuks: mida väiksem on energia, seda kergemini ergastub molekul. Kui elektron on ergastatud, hüppab ta hõivamata MO-le. See võib näiteks aidata ära arvata, kas miski kiirgab valgust (luminestsents).

Küsimused ja vastused
K: Mis on molekulaarorbiit?
V: Molekulaarorbitaal (või MO) on matemaatiline funktsioon, mis kirjeldab elektroni lainelist käitumist molekulis. See selgitab, mis juhtub elektronidega, kui aatomid ühinevad molekulis ja võib öelda, kui suur on tõenäosus leida elektron mõnes konkreetses piirkonnas.
K: Kuidas keemikud koostavad molekulaarorbitaalide matemaatilisi mudeleid?
V: Keemikud koostavad molekuliorbitaalide matemaatilisi mudeleid tavaliselt aatomiorbitaalide kombineerimise teel. Kasutada võib ka molekuli iga aatomi hübriidorbitaale või muid aatomirühmade molekulaarorbitaale. Arvutid võivad töötada nende funktsioonidega.
K: Kuidas on kvantmehaanika seotud molekulide uurimisega?
V: Molekulaarorbitaalid võimaldavad keemikutel rakendada kvantmehaanikat molekulide uurimisel. Nad vastavad küsimustele selle kohta, kuidas aatomid molekulides koos püsivad, ning annavad ülevaate keemilistest ja füüsikalistest omadustest.
K: Mis on orbitaaldiagrammid?
V: Orbitaaldiagrammid on visuaalsed kujutised, mis näitavad, kus aatomi elektronid kõige tõenäolisemalt asuvad, lähtudes selle erinevatest ümaratest kujudest.
K: Kuidas toimivad hübriidorbitaalid?
V: Hübriidorbitaalid kombineerivad eri tüüpi aatomiorbitaale üheks uueks tüübiks, millel on unikaalsed omadused võrreldes selle komponentidega. Neid hübriidorbitaale kasutatakse sageli molekulaarorbitaalide matemaatiliste mudelite koostamisel.
K: Kuidas saavad arvutid aidata MO-de uurimisel?
V: Arvutid võivad aidata MO-de uurimisel, töötades nende funktsioonide kallal ja andes täpsemaid prognoose või selgitusi molekulide keemiliste ja füüsikaliste omaduste kohta.
Seotud artiklid
Autor
AlegsaOnline.com Mis on molekulaarorbitaal? Selgitus, omadused ja näited Leandro Alegsa
URL: https://et.alegsaonline.com/art/65861
Allikad
- nobelprize.org : Nobelprize.org