SN1-reaktsioon: nukleofiilne unimolekulaarne asendus — mehhanism ja näited
SN1-reaktsioon: selgitatakse nukleofiilset unimolekulaarset asendust, mehhanism, karbokatsiooni roll ja näited sekundaarsete/tertsiaarsete ühendite kohta.
NS1-reaktsioon on asendusreaktsioon orgaanilises keemias. "SN" tähistab nukleofiilset asendust ja "1" tähistab asjaolu, et kiirust määrav samm hõlmab ainult ühte molekuli (unimolekulaarne). Reaktsioon kulgeb tavaliselt läbi karbokatsiooni kui vaheühendi. Anorgaaniliste keemikute seas tuntakse NS1-reaktsiooni sageli dissotsiatiivse mehhanismina. Christopher Ingold jt pakkusid selle reaktsioonimehhanismi esmakordselt välja 1940. aastal.
Pildigalerii
3 Pildid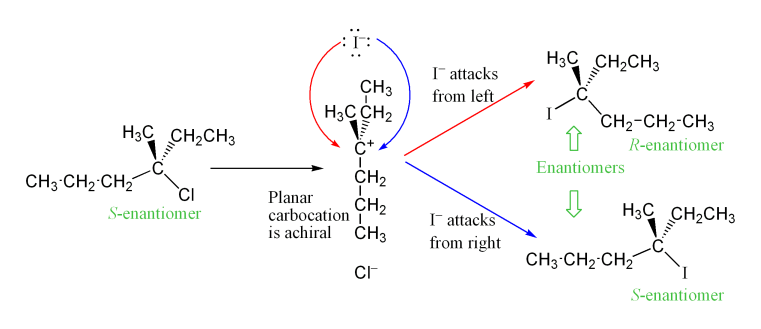
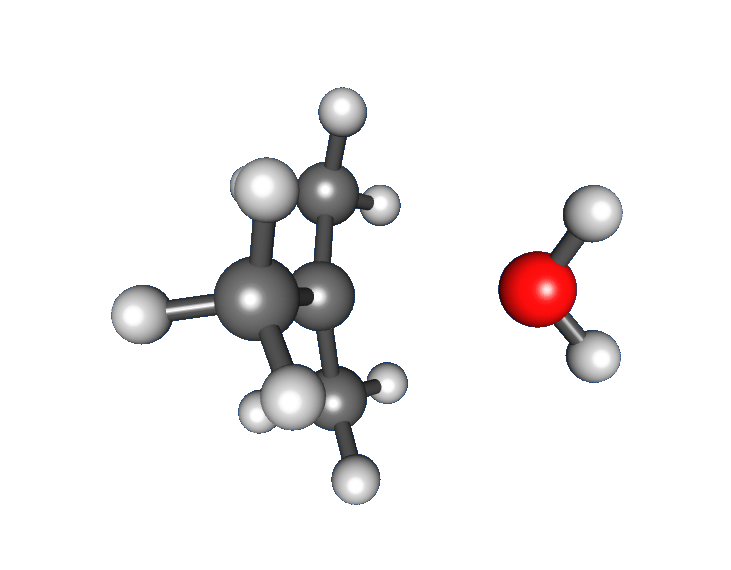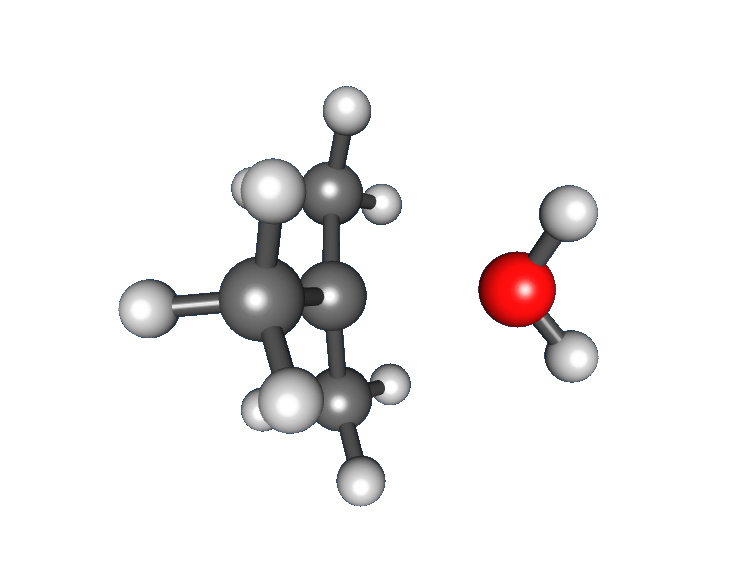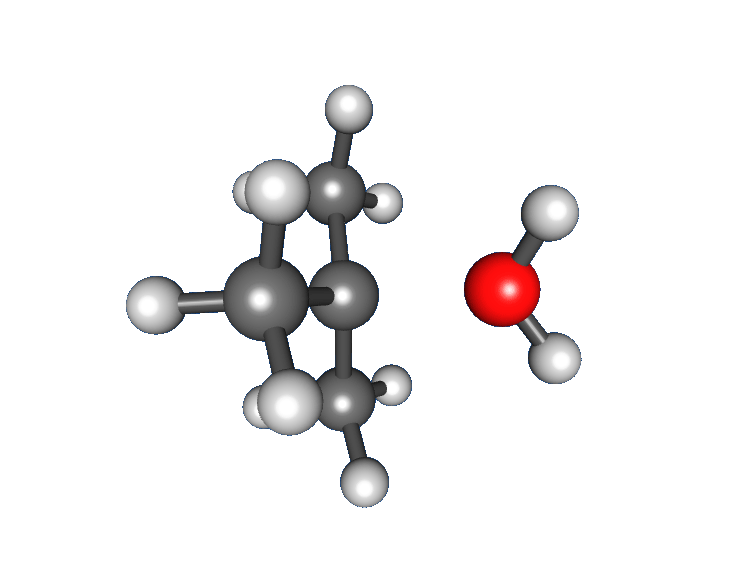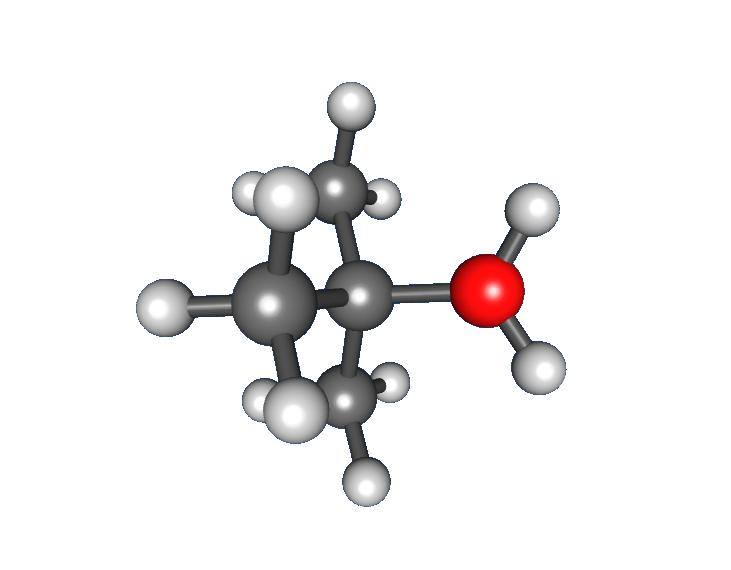

Mehhanism
SN1-mehhanism on kahest etapist koosnev:
- 1) Lahustuva rühma lahutus (lahkumine) — substraat (näiteks alküülhalogeniid või protonitud alkohol) kaotab leaving-grupi, moodustades karbokatsiooni. See samm on aeglasem ja seega kiirust määrav samm.
- 2) Nukleofiili lähenemine — vaba karbokatsioonile lisandub nukleofiil, moodustades lõpptoodangu.
Kuna kiirust määrav on ainult esimene samm, sõltub reaktsiooni kiirus ainult lähteaines oleva substradi kontsentratsioonist: rate = k[substrate]. Nukleofiili kontsentratsioon ei mõjuta kiirust oluliselt.
Faktorid, mis SN1-i soodustavad
- Karbokatsiooni stabiilsus: tertsiaarsed > sekundaarsed > primaarsed. Erinevad stabiliseerivad tegurid (induktne toime, hulgeneerimine, resonants) suurendavad SN1-liikumise tõenäosust.
- Resonantsi stabiilsus: bensüül- ja allyyl-substradid võivad SN1-reaktsioonis käituda kergemini, kuna karbokatsioonid on resonantsstabiilsed.
- Hea leaving‑grupp: tugevad leaving‑rühmad (nt halogeniidid, water pärast protoneerimist) soodustavad dissotsiatsiooni.
- Polaarne, protistaatne solvent: lahustid, mis stabiliseerivad ioone (nt vesi, alkoholid), soodustavad karbokatsiooni tekke ja seega SN1‑teed.
- Madalam nukleofiili tugevus: tugevad aluselised nukleofiilid eelistavad tihti bimolekulaarset NS2-reaktsiooni, mistõttu nõrgem-kuumem nukleofiil koos polaarse protistaatse keskkonnaga soosib SN1-i.
- Happelised tingimused alkoholide korral: alkohole tuleb esmalt protonida, et muuta –OH paremaks leaving‑grupiks; seetõttu kulgevad alkoholide asendused sageli happelises meediumis.
Stereokeemia ja vaheühendid
Kuna keskne karbokatsioon on sp2‑hübriidiseeritud ja plaaniline, võib nukleofiil lisanduda kahele poole võrdselt — seetõttu on oodata enamikul juhtudel otse racemise (racemise) tekke optiliselt aktiivsetest lähteainetest. Praktikas võib aga täheldada osalist säilimist või inverntsiooni, kui karbokatsiooni ja lahkuva rühma vahel on intiimne ionipaar (ion pair), mis piirab nukleofiili ligipääsu ühele küljelt.
Reaktsioonide näited ja rakendused
- Terts‑butüülkloriidi hüdrolüüs vees: t‑BuCl + H2O → t‑BuOH (solvolüüs), kus esineb SN1‑mehhanism ning kiirus sõltub ainult t‑BuCl kontsentratsioonist.
- Bensüül- ja allyülsubstradid: sekundaarsed bensüül‑ või allyül‑halogeniidid võivad SN1‑iga käituda kergemini kui tüüpilised primaarsed alkiilid, kuna karbokatsioon on resonantsiga stabiliseeritud.
- Alkoholide asendused happelistes tingimustes: alkoholi protonimine ja järgneva vee lahkumine moodustab karbokatsiooni, millele järgneb nukleofiilne lisandumine.
Spetsiifilised ilmingud ja erandid
Rearrangeerumised: sageli toimuvad 1,2‑hydride või alküül‑nihked, kui tekkinud karbokatsioon suudab ümber järjestada end stabiilsema karbokatsiooni suunas (näiteks 2° → 3°), mis muudab saadused ootamatuks võrreldes lihtsa asendusega.
Naudingud keemilises sünteesis: SN1‑teed kasutatakse teadlikult siis, kui soovitakse kasutada resonantsstabiilseid või tertsiaarseid keskkondi ning kui nukleofiilina on lahusti ise (solvolüüs) või nõrk nukleofiil.
Tõendid ja ajalugu
SN1‑mehhanismi toe andsid kiirust määravad eksperimendid, karbokatsioonide tuvastamine ja reagendid, mis karbokatsioone tabavad (trapping), samuti sõltuvus lahusti polaarsusest ja leaving‑rühma paremuse mõjust kiirusele. Nagu mainitud, oli selle mehhanismi algne kirjeldus Christopher Ingoldi ja kolleegide töö tulemus 1940. aastatel (reaktsioonimehhanism).
Kokkuvõtlikult: SN1 on unimolekulaarne nukleofiilne asendus, mida iseloomustab karbokatsiooni vaheühendi teke, kiiruse sõltumine ainult substradist, soodustamine polaarsetes protistaatsetes lahustites ning kalduvus racemise ja rearrangeerumiste poole. Primaarsete alküülhalogeniidide puhul on tavaline alternatiiv NS2-reaktsioon.
Mehhanism
Näide NS1-reaktsioonimehhanismiga toimuva reaktsiooni kohta on tert-butüülbromiidi hüdrolüüs veega tert-butüülalkoholi moodustamiseks:
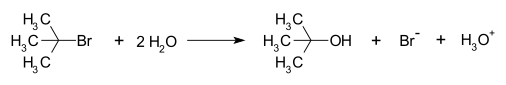
See NS1-reaktsioon toimub kolmes etapis:
- tert-butüülkarbokatsiooni moodustumine lahkumisrühma (bromiidi anioon) eraldamise teel süsiniku aatomist; see etapp on aeglane ja pöörduv.
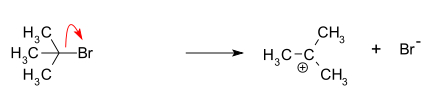
- Nukleofiilne rünnak: karbokatioon reageerib nukleofiiliga. Kui nukleofiil on neutraalne molekul (st lahusti), on reaktsiooni lõpuleviimiseks vaja kolmandat sammu. Kui lahusti on vesi, on vaheproduktiks oksooniumioon. See reaktsioonietapp on kiire.

- Deprotoneerimine: Prootoniseeritud nukleofiili prootoni eemaldamine vee poolt, mis toimib alusena, moodustades alkoholi ja hüdroniumiooni. See reaktsioonietapp on kiire.
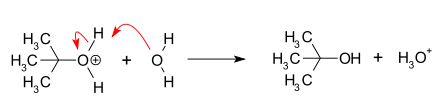
Kuna esimene samm on kitsaskoht või "kiirust määrav samm", liigitavad keemikud kogu reaktsioonimehhanismi NS1-ks. Selleks etapiks on vaja ainult ühte molekuli.
Reaktsiooni ulatus
Mõnikord võib molekul reageerida kas NS1- või NS2-mehhanismi abil. NS1-mehhanism võidab selle võistluse, kui keskne süsinikuaatom on ümbritsetud mahukate rühmadega, sest sellised rühmad takistavad steriilselt NS2-reaktsiooni. Lisaks suurendavad mahukad substituendid keskse süsiniku aatomi juures karbokatsiooni moodustumise kiirust, sest see vähendab steriilset pinget. Saadud karbokatiooni stabiliseerib ka nii induktiivne stabiliseerimine kui ka lisatud alküülrühmadest tulenev hüperkonjugatsioon. Hammond-Leffleri postulaat ütleb, et ka see suurendab karbokatsiooni moodustumise kiirust. NS1-mehhanism domineerib seega tertsiaarsete alküülkeskuste juures toimuvates reaktsioonides ja seda täheldatakse ka sekundaarsete alküülkeskuste juures nõrkade nukleofiilide juuresolekul.
Näide NS1-meetodil kulgeva reaktsiooni kohta on 2,5-dikloro-2,5-dimetüülheksaani süntees vastavast dioolist kontsentreeritud soolhappega:
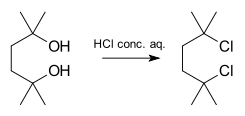
Kuna alfa- ja beetasubstitutsioonid suurenevad lahkuvate rühmade suhtes, suunatakse reaktsioon NS2-lt NS1-le.
Stereokeemia
Reaktsiooni kiirust piiravas etapis moodustuv karbokatioonivaheline produkt on trigonaalse planaarse molekulaargeomeetriaga sp-hübriidne2 süsinik. See võimaldab nukleofiilse rünnaku jaoks kaks erinevat teed, üks mõlemal pool planaarset molekuli. Kui kumbki tee ei ole eelistatud, kasutatakse neid kahte teed võrdselt, saades enantiomeeride raseemilise segu, kui reaktsioon toimub stereotsentris. Seda illustreerib allpool S-3-kloro-3-metüülheksaani NS1-reaktsioon jodiidiooniga, mille tulemuseks on 3-jodo-3-metüülheksaani raseemiline segu:
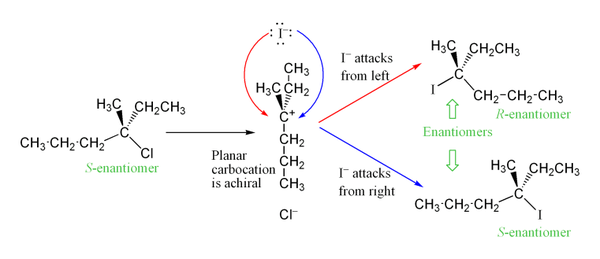
Siiski võib täheldada ühe stereoisomeeri ülejääki, kuna lahkuv rühm võib jääda lühikeseks ajaks karbokatsiooni vaheühendi lähedusse ja blokeerida nukleofiilset rünnakut. See on väga erinev NS2-mehhanismist, mis ei sega produkti stereokeemiat (stereospetsiifiline mehhanism). NS2-mehhanism inverteerib alati molekuli stereokeemiat.
Kõrvalreaktsioonid
Kaks tavalist kõrvalreaktsiooni on eliminatsioonireaktsioonid ja karbokatsioonide ümberpaiknemine. Kui reaktsioon viiakse läbi soojades või kuumades tingimustes (mis soodustavad entroopia suurenemist), on tõenäoline, et E1 eliminatsioon domineerib, mis viib alkeeni moodustumiseni. Madalamatel temperatuuridel on NS1- ja E1-reaktsioonid konkureerivad reaktsioonid. Seega on raske eelistada ühte neist teise suhtes. Isegi kui reaktsioon viiakse läbi külmalt, võib tekkida mõni alkeen. Kui NS1-reaktsiooni püütakse läbi viia, kasutades tugevalt aluselist nukleofiili, näiteks hüdroksiid- või metoksiidiooni, moodustub alkeen jälle, seekord E2 eliminatsiooni kaudu. See kehtib eriti siis, kui reaktsiooni kuumutatakse. Lõpuks, kui karbokatiooni vaheprodukt võib ümber korraldada stabiilsemaks karbokatiooniks, siis tekib pigem stabiilsemast karbokatioonist saadud toode kui lihtne asendusprodukt.
Lahusti mõju
Lahustid muudavad reaktsioonikiirust. Kuna NS1-reaktsioon hõlmab ebastabiilse karbokatsiooni vaheühendi moodustumist kiirust määravas etapis, kiirendab reaktsiooni kõik, mis seda aitab. Tavapärased lahustid on nii polaarsed (et stabiliseerida ioonseid vaheühendeid üldiselt) kui ka protilised (et lahustuv rühm solvateeruks). Tüüpilised polaarsed protilised lahustid on vesi ja alkoholid, mis toimivad ka nukleofiilidena.
Y-skaala korreleerib mis tahes lahusti (k) ja standardlahusti (80% v/v etanool/vesi) solvolüüsi reaktsioonikiirused (k0) läbi
log ( k k ) 0= m Y {\displaystyle \log {\left({\frac {k}{k_{0}}}\right)}=mY\,} 
kusjuures m on reaktiivikonstant (m = 1 tert-butüülkloriidi puhul),
- Y lahusti parameeter ja
- k0 on reaktsioonikiirus, kasutades lahustina 80% etanooli (mõõdetuna mahu järgi).
Näiteks 100% etanool annab Y = -2,3, 50% etanool vees Y = +1,65 ja 15% kontsentratsioon Y = +3,2.
Küsimused ja vastused
K: Mida tähendab "SN" SN1-reaktsioonis?
V: "SN" tähistab nukleofiilset asendust.
K: Mida tähistab "1" SN1-reaktsioonis?
V: "1" tähistab asjaolu, et kiirust määravasse sammu on kaasatud ainult üks molekul (unimolekulaarne).
K: Millist tüüpi reaktsioon on SN1-reaktsioon?
V: SN1 on asendusreaktsioon.
K: Milline on SN1-reaktsioonis osalev vaheprodukt?
V: SN1-reaktsioonis osaleb karbokatsioonide vaheühend.
K: Millistel tingimustel toimuvad tavalised SN1-reaktsioonid?
V: Tavalised SN1-reaktsioonid toimuvad sekundaarsete või tertsiaarsete alküülhalogeniididega tugevalt aluselistes tingimustes või sekundaarsete või tertsiaarsete alkoholidega tugevalt happelistes tingimustes.
K: Milline alternatiivne reaktsioon toimub primaarsete alküülhalogeniididega?
V: Primaarsete alküülhalogeniididega toimub alternatiivne SN2-reaktsioon.
K: Kes pakkus esimesena välja SN1-reaktsiooni mehhanismi ja millisel aastal?
V: Christopher Ingold et al. pakkusid SN1-reaktsiooni mehhanismi esmakordselt välja 1940. aastal.
Seotud artiklid
Autor
AlegsaOnline.com SN1-reaktsioon: nukleofiilne unimolekulaarne asendus — mehhanism ja näited Leandro Alegsa
URL: https://et.alegsaonline.com/art/91288
Allikad
- sciteclibrary.ru : SciTecLibrary