Creutzfeldt–Jakobi tõbi (CJD) — prioonhaigus, sümptomid ja prognoos
Creutzfeldt–Jakobi tõbi (CJD) — prioonhaigus: sümptomid, diagnoos, ravi puudumine ja prognoos. Loe riskidest, varajastest tunnustest ja mida teada haiguse kulust.
Creutzfeldt-Jakobi tõbi (hääldatakse KROITS-felt YAH-kohb) ehk CJD on neuroloogiline haigus. See on degeneratiivne (aja jooksul halveneb); seda ei saa ravida ja see põhjustab alati surma. CJD-d nimetatakse mõnikord "hullu lehma tõve" (veiste spongioosse entsefalopaatia ehk BSE) inimvormiks. BSE on tegelikult Creutzfeldt-Jakobi tõve ühe harvaesineva tüübi põhjustaja; need kaks ei ole üks ja sama haigus.
CJD on põhjustatud nakkusetekitajast, mida nimetatakse priooniks. Prioonid on valkud, mis on valesti volditud. Prioonid teevad iseendast koopiaid, muutes õigesti volditud valke vääralt volditud kujuliseks. CJD põhjustab ajukoe väga kiiresti ebaterveks muutumist. Kuna haigus hävitab aju, tekivad ajus augud. Aju tekstuur muutub ja muutub nagu köögi käsn.
Pildigalerii
6 Pildid




Mis juhtub ajus ja mis on prioonid?
Prioonhaigused tekivad siis, kui normaalselt volditud ajuvalk (PrPC) omandab ebanormaalse, kergesti agregaatuvate omadustega kuju (PrPSc). Need valed voldid põhjustavad teiste samade valkude vääravoldumist, moodustades lahustumatuid ladestusi. See viib neuronite hävimiseni, gliosini ja spongioosse ehk "vahatud" ajukoetüki tekkeni. Erinevad prioonhaigused (sh CJD, Kuru, BSE, fataalne perekondlik unetus) on samale mehhanismile tuginevad, kuid kliinilised tunnused ja haiguse kulg võivad erineda.
Liigid ja ülekandumine
- Sporadiline CJD — kõige levinum vorm (umbes 85–90% juhtudest). Ilmneb ilma selge põhjuseta, tavaliselt vanuses 60–65 eluaastat.
- Perekondlik (familiar) CJD — tekib PRNP-geeni mutatsiooni tõttu ja pärandub. Haigus võib alata nooremas eas ning esineda perekondades.
- Iatrogeenne CJD — retsiprokaalne ülekandumine meditsiiniliste protseduuride kaudu (varasemalt on dokumenteeritud juhtumeid seostatuna saastunud neurokirurgiliste instrumentide, dura mater'i implantaadi või kasvuhormooni siirdamisega). Tänapäeval on risk oluliselt väiksem tänu pingutustele ravi- ja steriliseerimisprotseduuride osas.
- Variant-CJD (vCJD) — seotud BSE-ga; esineb peamiselt noorematel patsientidel ja avaldub sageli algsete psühhiaatriliste sümptomitega ning pikema haigusajalooga kui sporadiline CJD.
Sümptomid
CJD sümptomid arenevad tavaliselt kiiresti (nädalate kuni kuude jooksul). Kõige sagedasemad on:
- Mälukaotus ja kiire kognitiivne langus (dementsuse kiire areng)
- Erinevad koordinatsiooni- ja tasakaaluhäired (ataksia)
- Ärevus, depressioon, käitumismuutused ja mõnikord psühhiaatrilised sümptomid (eriti variant-CJD puhul)
- Liikumishäired: lihastõmblused (miokloonused), jäikus, ebaühtlane lihasjõud
- Häired kõne ja neelamise funktsioonis ning lõpufaasis teadvusekaotus ja täielik puudega seisund
Diagnoos
Diagnoos põhineb kliinilisel pildil, neuroloogilisel uuringul ja abiuuringutel. Levinumad diagnostilised leiud:
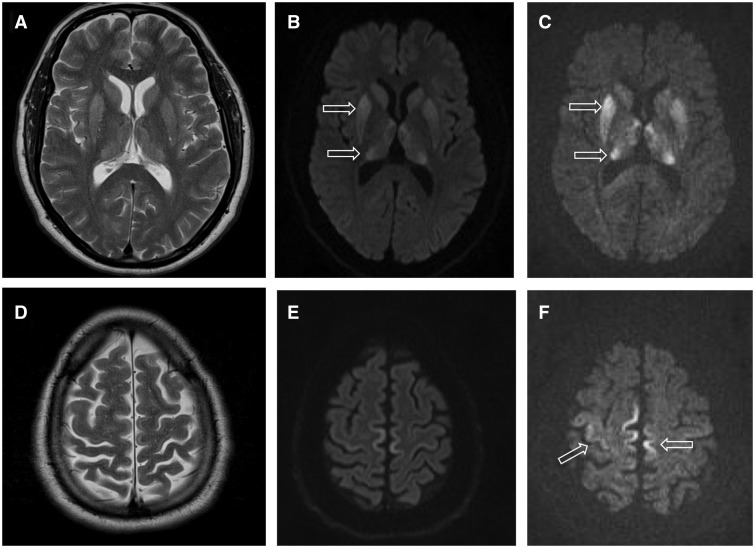
- MRI: iseloomulikud DWI/FLAIR muutused (nn taastusribi ehk cortical ribboning, ning kauduataalsed tuumad) aitavad diagnoosi toetada.
- EEG: mõnel juhul leiab perioodilisi terav-lainemustreid (periodic sharp wave complexes), kuid see ei ole alati olemas.
- CSF (seljaajuvedelik): 14-3-3 valk võib olla positiivne, kuid see ei ole spetsiifiline. Tänapäeval RT-QuIC (real-time quaking-induced conversion) on tundlik ja spetsiifiline test, mis tuvastab priooni olemasolu.
- Definitiivne diagnoos tehakse tavaliselt aju biopsia või post-mortem uuringu (autopsia) abil, kus nähakse spongioosseid muutusi ja prioonvalgu ladestusi.
Ravi ja prognoos
Praegu puudub CJD-le raviv teraapia, mis peataks või pööraks haiguse. Ravi on toetav ja sümptomaatiline: valu- ja ärevuse leevendamine, toidu ja vedeliku manustamise korraldus, hingamistoetuse vajaduse hindamine ning palliatiivne hooldus. Mõned eksperimentaalsed ravivõtted on uurimisel, kuid kuni kinnitatud efektiivset ravimit pole, jääb CJD prognoos väga halbaks.
Sporadilise CJD korral on prognoos tavaliselt raske: sümptomite ilmnemisest kuni surmani möödub keskmiselt mitu kuud; enamik patsiente sureb aasta jooksul. Variant-CJD-l võib haigus kulgeda pikemalt ja patsientide vanus on sageli madalam.
Ennetus ja nakkuskontroll
Prioonid on tavapärase desinfitseerimise ja steriliseerimise suhtes erakordselt resistentsed. Sellepärast on meditsiinilistes olukordades, kus on risk priooniülekandeks (neurokirurgia, teatud implantaadid), rangelt järgitud spetsiaalsed desinfitseerimisprotokollid ja piirangud. Riiklikud tervishoiuametid annavad juhised instrumentide töötlemiseks ja riski vähendamiseks. Samuti kehtivad vere- ja elundidoonorluse piirangud, et vähendada vCJD-ga seotud riske.
Edaspidi on oluline pidev haiguste järelevalve ja teadlikkus (raporteerimine ja epidemioloogiline jälgimine), et avastada ja ennetada uusi juhtumeid ning piirata iatrogeenseid edasikandumisi.
Lisamärkus
Kuigi CJD on harvaesinev (eelarvudeks tavaliselt ~1–2 juhtumit miljoni elaniku kohta aastas), on haiguse ränk kulg ning nakkuslik olemus teinud temast olulise avaliku tervise küsimuse, eriti seoses loomsete prioonhaiguste (nt BSE) võimaliku mõjuga inimestele. Kui kahtlustatakse CJD-d, on oluline pöörduda kiiresti spetsialisti poole ja järgida kohalikke juhiseid nakkuskontrolliks.
CJD tüübid ja põhjused
CJD tüübid on järgmised:
- variant (vCJD):
Seda tüüpi CJD võib põhjustada toidu söömine, mis sisaldab prioone, näiteks BSE-haigusega lehmade liha ("hullu lehma tõbi"). See on siiski väga haruldane CJD põhjus.
- sporaadiline (sCJD):
See on kõige levinum CJD tüüp. 85% CJD juhtudest on sporaadiline CJD. Keegi ei tea, mis põhjustab sCJD-d; see näib juhtuvat juhuslikult.
- perekondlik (fCJD):
Enamik ülejäänud 15% CJD juhtudest on perekondlik CJD. See on CJD vorm, mis esineb perekondlikult.
- iatrogeenne:
See CJD vorm on tavaliselt põhjustatud meditsiinilisest protseduurist, mille käigus inimene saab verd või kude mõnelt CJD-ga inimeselt. Näiteks võib inimene saada iatrogeense CJD, kui ta saab vereülekannet või sarvkesta siirdamist inimeselt, kellel on CJD.
Märgid ja sümptomid
Esimene CJD sümptom on dementsus, mis süveneb väga kiiresti. dementsus põhjustab mälukaotust, isiksuse muutusi ja hallutsinatsioone.
Muud tavalised psüühilised sümptomid on järgmised:
- Ärevus
- Depressioon
- Paranoia
- Obsessiiv-kompulsiivsed sümptomid
- Psühhoos
CJD füüsilised sümptomid on sageli järgmised:
- Raskused rääkimisega
- Närvilised liigutused (müokloonus)
- Tasakaaluprobleemid (ataksia)
- Probleemid kõndimisega
- Värisemine või jäigaks jäämine
- Nägemisprobleemid
- neelamisraskused, mis võivad muuta söömise raskeks või võimatuks.
- Köhaprobleemid, mis võivad põhjustada kopsupõletikku.
- liigutused, mida patsient ei suuda kontrollida (düskineesia).
Enamik CJDsse haigestunutest sureb kuue kuu jooksul pärast esimeste sümptomite ilmnemist. Sageli surevad nad kopsupõletikku, mis on põhjustatud köhaprobleemidest. Umbes 15% patsientidest elab kaks või enam aastat. Mõned patsiendid on elanud 4-5 aastat peamiselt vaimsete sümptomitega, kuni haigus süveneb ja põhjustab rohkem füüsilisi sümptomeid. Kui see juhtub, surevad inimesed tavaliselt aasta jooksul.
CJD sümptomid on põhjustatud üha enamate aju närvirakkude surmast. Kui teadlased vaatavad mikroskoobi all CJD patsiendi ajukude, näevad nad palju pisikesi auke, kus on surnud terveid närvirakkude piirkondi.
Diagnoos
Arstid võivad kahtlustada CJD-d, kui inimesel on teatud sümptomid. Näiteks dementsus süveneb tavaliselt aeglaselt. Väga kiiresti süvenev dementsus on ebatavaline. Koos selliste sümptomitega nagu tõmblused, võivad need sümptomid viidata võimalikule CJD-le.
Seejärel saab teha teste, mis näitavad, kas isikul on CJD. Need testid hõlmavad järgmist:
- Elektroentsefalograafia (EEG): See test näitab elektrilist aktiivsust ajus. Arst on sageli võimeline nägema EEG-s muutusi, mis on tavalised CJD-ga inimestel. EEG-l ilmnevate muutuste tüüp sõltub sellest, millist tüüpi CJD on patsiendil ja kui kaugel on tema haigus.
- Lumbaalpunktsioon (spinaalpunktsioon): See test võimaldab uurida seljaaju vedelikku (aju ja seljaaju ümbritsev vedelik), otsides spetsiifilist valku ("14-3-3 valk").
- Aju magnetresonantstomograafia: Test, milles kasutatakse väga tugevat magnetit, et teha ajust pilte.
- Biopsia: Biopsia tegemiseks võtab kirurg nõelaga kehast väikese koetüki, et arstid saaksid seda mikroskoobi all vaadata. vCJD-d saab diagnoosida mandlite biopsia abil. Kõigi teiste CJD liikide puhul on aju biopsia ainus võimalus kindlalt öelda, kas inimesel on CJD. Kuna aga aju biopsia võib põhjustada ajukahjustusi, ei tehta tavaliselt aju biopsiat, kui muud testid on juba näidanud, et isikul on tõenäoliselt CJD.

Töötlemine
2016. aasta seisuga ei ole olemas ravi, mis raviks CJD-d või isegi aeglustaks selle mõju. Ravimeetodite leidmiseks tehakse palju katseid.
Tänapäeval on CJD ainus ravi ravimid, mis ravivad haiguse sümptomeid ja aitavad patsientidel end paremini tunda. Näiteks võib patsientidele, kellel on krambid, anda krambivastaseid ravimeid. Bensodiasepiinide abil võivad lihastõmblused harvemini tekkida.
Patsiendid võivad valida ka meditsiinilisi protseduure, et aidata halbu sümptomeid leevendada. Näiteks võib CJD põhjustada nii palju neelamisraskusi, et inimene ei saa süüa. Mõned CJDga inimesed otsustavad, et neile pannakse sond, kui nad ei saa enam süüa. See on toru, mis läheb maosse, nii et spetsiaalset vedelikku saab anda otse maosse, et anda inimesele toitu.
Seotud leheküljed
- Prion
- Prionihaigus
- Terminaalne haigus
Küsimused ja vastused
K: Mis on Creutzfeldt-Jakobi tõbi?
V: Creutzfeldt-Jakobi tõbi (CJD) on neuroloogiline haigus, mis on degeneratiivne, ravimatu ja alati surmaga lõppev.
K: Kas CJD-d on võimalik ravida?
V: Ei, CJD-d ei ole võimalik ravida.
K: Miks nimetatakse CJD-d mõnikord "hullu lehma tõve" inimvormiks?
V: CJD-d nimetatakse mõnikord "hullu lehma tõve" inimvormiks, sest veiste spongioosset entsefalopaatiat (BSE), mis on ühe harvaesineva CJD tüübi põhjustaja, tuntakse üldiselt "hullu lehma tõvena".
K: Mis on CJD põhjus?
V: CJD-d põhjustab nakkusetekitaja, mida nimetatakse priooniks, mis on valk, mis on valesti volditud ja võib teha koopiaid iseendast, muutes õigesti volditud valke valkudeks, mis on valesti viltu.
K: Mis juhtub CJD puhul ajukoes?
V: CJD põhjustab ajukoe väga kiiresti ebaterveks muutumist, mille tagajärjel tekivad ajus augud ja aju tekstuur muutub nagu köögipesu.
K: Kas BSE on sama haigus kui CJD?
V: Ei, BSE ei ole sama haigus kui CJD; see on tegelikult ühe harvaesineva CJD tüübi põhjus.
K: Kuidas põhjustavad prioonid CJD-d?
V: Prioonid põhjustavad CJD-d, kuna need volditakse valesti ja valmistavad endast koopiaid õigesti volditud valkude arvelt ajus. Selle tulemuseks on terve ajukoe hävimine ja haigusele iseloomulike aukude teke.
Seotud artiklid
Autor
AlegsaOnline.com Creutzfeldt–Jakobi tõbi (CJD) — prioonhaigus, sümptomid ja prognoos Leandro Alegsa
URL: https://et.alegsaonline.com/art/24152
Allikad
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"